
Polycystic Kidney Disease: The Genetic Condition That Fills Kidneys With Cysts
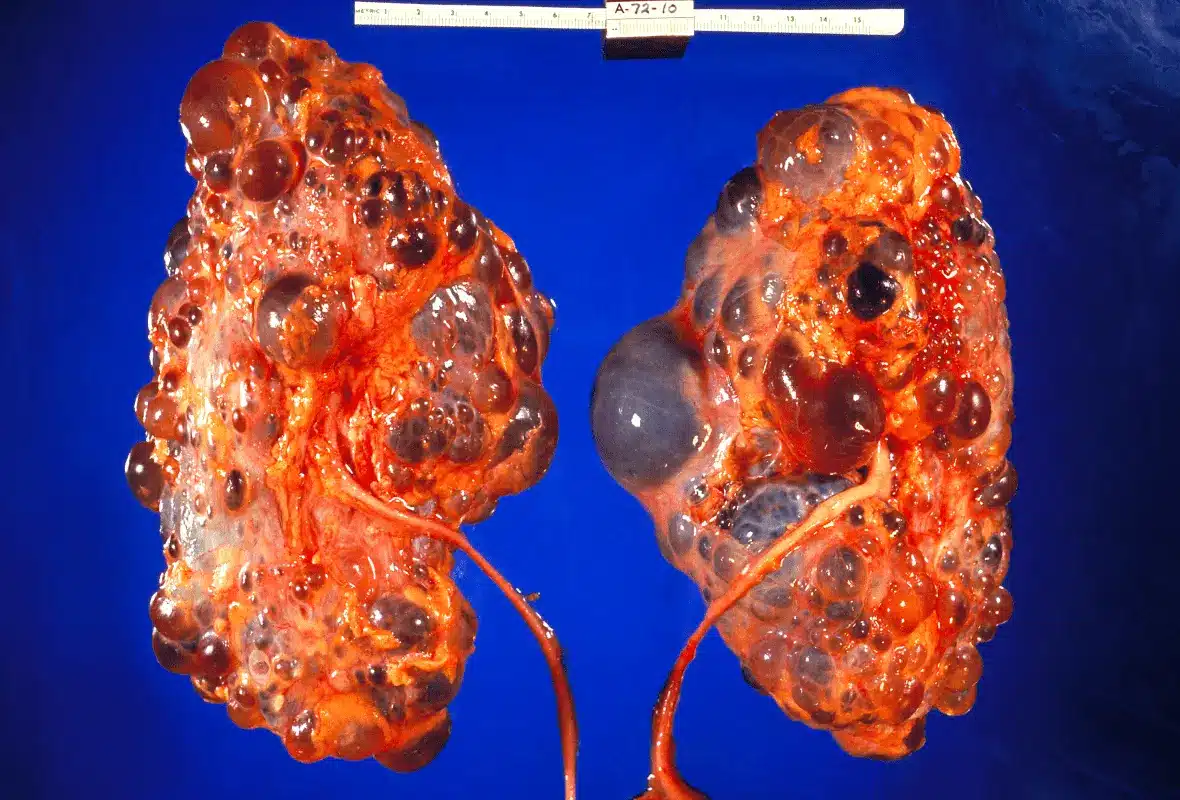
Most genetic kidney conditions damage the kidneys by attacking their filtering units from outside. Polycystic kidney disease works differently. It builds a slow-growing problem from within — filling the kidneys with fluid-filled sacs that grow steadily over decades until the organ can no longer function.
Polycystic kidney disease is the most common inherited kidney disorder in the world. It affects roughly 12.5 million people globally, regardless of sex, ethnicity, or location. As hundreds and then thousands of cysts form and expand inside the kidneys, they destroy surrounding healthy tissue and reduce the kidneys’ ability to filter blood.
Polycystic kidney disease genetic cysts kidneys represent a condition where the damage is encoded in DNA from the very beginning of life. However, understanding how the disease works, how it progresses, and what treatments are available gives patients and families a real ability to slow that progression and protect quality of life for decades.
Quick Answer: What Is Polycystic Kidney Disease?
Polycystic kidney disease — commonly called PKD — is an inherited genetic disorder. Abnormal fluid-filled cysts develop and grow inside both kidneys over time. These cysts arise from the renal tubules — tiny channels that process filtered fluid within each nephron. Over time, the cysts multiply, enlarge, and press on surrounding healthy kidney tissue.
PKD exists in two main genetic forms. Autosomal dominant PKD — called ADPKD — is the most common form and typically affects adults. Autosomal recessive PKD — called ARPKD — is far rarer and mainly affects newborns and children. Furthermore, PKD is the fourth most common cause of kidney failure worldwide, responsible for roughly 5 to 10% of all cases requiring dialysis or transplantation.
How Cysts Form and Grow in PKD
The Role of the Primary Cilium
Understanding polycystic kidney disease genetic cysts kidneys requires a picture of how cysts form and why they grow so persistently. In a healthy kidney, cells lining the renal tubules control fluid secretion, cell growth, and tubule structure. This control depends on a tiny hair-like projection on each cell called the primary cilium. It detects fluid flow and sends regulatory signals into the cell.
In PKD, genetic mutations disrupt the proteins that make the primary cilium work correctly. As a result, the normal regulatory signals break down. Tubular cells begin to grow abnormally and secrete fluid into the tubule instead of absorbing it. Consequently, small outpouchings form along the tubule wall and fill with fluid. These outpouchings break away from the original tubule and form independent, self-expanding cysts.
Why Cysts Keep Growing
Once a cyst forms, moreover, it develops its own growth programme. The cells lining the cyst wall keep growing and secreting fluid. This process is driven by dysregulated signalling pathways — particularly the mTOR pathway and the cyclic AMP pathway. Consequently, the cyst expands steadily regardless of its impact on surrounding tissue.
Over decades, individual cysts grow from microscopic to centimetre-sized structures. Furthermore, the total kidney volume — a key measure of disease progression — increases steadily as more cysts accumulate. As a result, the kidneys can eventually grow to many times their normal size.
Types of Polycystic Kidney Disease
Autosomal Dominant PKD
Autosomal dominant PKD is by far the most common form, accounting for roughly 90% of all PKD cases. It follows an autosomal dominant inheritance pattern. This means that inheriting just one mutated copy of the disease gene from either parent causes the condition. Consequently, each child of an affected parent has a 50% chance of inheriting the mutated gene.
ADPKD results from mutations in one of two genes. Mutations in the PKD1 gene on chromosome 16 account for roughly 75 to 80% of ADPKD cases and generally cause more severe disease with earlier kidney failure. Mutations in the PKD2 gene on chromosome 4 account for the remaining 20 to 25% of cases and typically follow a milder and slower course. Furthermore, roughly 10% of ADPKD cases arise from new mutations that appear for the first time in the affected person without being inherited from a parent.
ADPKD typically remains silent through childhood and early adulthood. Symptoms generally emerge in the third to fifth decade of life. Consequently, many people reach their forties or fifties before diagnosis, by which time significant cyst burden has already built up.
Autosomal Recessive PKD
Autosomal recessive PKD is far rarer, affecting roughly one in 20,000 live births. It requires the inheritance of two mutated copies of the PKHD1 gene — one from each parent. Consequently, both parents must carry the mutation for a child to be affected. Each child of two carriers has a 25% chance of developing the condition.
Unlike ADPKD, moreover, ARPKD primarily affects the kidney collecting ducts and the liver bile ducts at the same time. Consequently, liver scarring — called congenital hepatic fibrosis — is a consistent feature of ARPKD and can itself cause serious complications.
ARPKD presents much earlier and more severely than ADPKD. The most severe cases show massively enlarged kidneys on prenatal ultrasound before birth. Furthermore, severely affected newborns may experience breathing difficulties at birth because massively enlarged kidneys compress the developing lungs in the womb. Consequently, ARPKD carries a much higher early risk of death than ADPKD.
What Causes Polycystic Kidney Disease?
PKD1 and PKD2 Gene Mutations
Polycystic kidney disease genetic cysts kidneys originate entirely from inherited or new genetic mutations. The PKD1 gene produces a protein called polycystin-1. The PKD2 gene produces polycystin-2. These two proteins work together in the primary cilium of renal tubular cells. Together, they control calcium signalling, cell polarity, and cell growth.
Consequently, when either protein is absent or non-functional due to a mutation, the primary cilium loses its ability to regulate the cell. Cyst formation then begins. PKD1 mutations cause more severe disease because polycystin-1 plays a broader role across more signalling pathways. In contrast, PKD2 mutations affect a more limited set of calcium channel functions. Therefore, PKD1 mutations lead to kidney failure — typically in the mid-fifties — roughly a decade earlier than PKD2 mutations.
The Two-Hit Hypothesis
An important concept in understanding PKD is the two-hit hypothesis. Each kidney cell carries two copies of the relevant PKD gene — one from each parent. In ADPKD, one copy carries a mutation from birth. However, cyst formation needs the second, working copy to also develop a mutation — a random error that occurs in an individual tubular cell during normal cell division.
This second event happens randomly in individual cells throughout life. Consequently, only a small number of the millions of tubular cells in the kidney develop into cysts at any time. This explains why PKD progresses over decades rather than causing immediate kidney failure — new cysts form gradually as these random errors accumulate in individual cells over many years.
Genetic Modifiers
Beyond the primary PKD1 and PKD2 mutations, several genetic factors influence how severe the disease becomes. Mutations that completely destroy protein function in PKD1 produce more severe disease than those that only partially reduce protein activity. Furthermore, certain modifier genes affecting the mTOR and Wnt signalling pathways influence the rate of cyst growth. Consequently, two people with identical PKD1 mutations may experience significantly different rates of disease progression.
Symptoms of Polycystic Kidney Disease
Early Symptoms in ADPKD
Polycystic kidney disease genetic cysts kidneys typically cause no symptoms in early adulthood despite the presence of growing cysts. The kidneys have enormous reserve — a person can lose more than half their nephrons before any measurable drop in eGFR occurs. Consequently, most people with ADPKD are diagnosed in their thirties or forties either because of a family history prompting screening or because symptoms finally appear.
The earliest and most common symptom is high blood pressure. It affects roughly 60% of ADPKD patients before any measurable drop in kidney function occurs. High blood pressure in ADPKD arises because enlarging cysts press on renal blood vessels, activating a hormonal system that raises blood pressure. Furthermore, uncontrolled blood pressure speeds up cyst growth and kidney function decline, creating a harmful cycle.
Flank pain — pain in the side between the ribs and hip — is another early and common symptom. It arises from the physical effect of enlarging kidneys stretching the renal capsule and pressing on nearby structures. In addition, sudden and severe flank pain may signal bleeding into a cyst or the passage of a kidney stone. Both occur more frequently in PKD than in the general population.
Progressive Symptoms
As total kidney volume increases and eGFR begins to fall, symptoms of kidney decline appear. Blood in the urine occurs in roughly 35 to 50% of ADPKD patients at some point. It results from cyst rupture into the urinary system or from kidney stone passage. Moreover, urinary tract infections are more common in PKD because stagnant fluid within cysts gives bacteria a place to grow.
Abdominal fullness and a visibly enlarged abdomen develop as the kidneys grow to extraordinary sizes. In advanced ADPKD, moreover, each kidney can reach the size of a football — compared with the normal weight of roughly 150 grams. Consequently, the massively enlarged kidneys press on and push aside nearby abdominal organs, causing significant discomfort and digestive symptoms.
Extra-Renal Manifestations
PKD is not solely a kidney disease. Polycystic kidney disease genetic cysts kidneys represent only one dimension of a wider condition affecting multiple organs. Furthermore, these effects outside the kidneys add significantly to the overall burden of disease and need active monitoring.
Liver cysts are the most common non-kidney finding in ADPKD, affecting roughly 80 to 90% of patients by age 60. In most cases, liver cysts do not affect liver function. However, massive polycystic liver disease can cause severe abdominal swelling, pain, and pressure on nearby structures. This occasionally needs surgical treatment.
Brain aneurysms — weaknesses in the walls of brain blood vessels — affect roughly 8 to 12% of people with ADPKD. This rate is four to five times higher than in the general population. Consequently, rupture of a brain aneurysm causing bleeding around the brain is one of the most serious non-kidney complications of ADPKD. Furthermore, screening for brain aneurysms is recommended for ADPKD patients with a family history of aneurysm rupture or bleeding stroke.
Additional non-kidney findings include heart valve abnormalities — particularly mitral valve prolapse affecting roughly 25% of patients — widening of the aortic root, pancreatic cysts, seminal vesicle cysts, and abdominal wall hernias. Moreover, bowel diverticula occur more frequently in people with ADPKD than in the general population.
How Doctors Diagnose Polycystic Kidney Disease
Imaging Studies
Renal ultrasound is the first-line diagnostic tool for PKD. It is non-invasive, widely available, and accurately identifies kidney cysts in most cases. Age-specific criteria exist for ADPKD — finding three or more cysts in both kidneys in a person under 40 with a family history of ADPKD provides enough diagnostic confidence in most cases. Furthermore, the total kidney volume measured on ultrasound or MRI provides important information about the likely rate of future progression.
MRI provides more precise measurement of total kidney volume than ultrasound. Consequently, MRI is the preferred tool for monitoring disease progression in clinical trials and for patients being considered for targeted drug treatment. CT scanning provides similar detail to MRI but exposes patients to radiation and contrast agents. Therefore, CT is generally saved for specific situations such as suspected cyst infection, bleeding, or concern about cancer.
Genetic Testing
Genetic testing for PKD1 and PKD2 mutations plays an important role in certain situations. It helps confirm the diagnosis in people with no family history and unusual imaging findings. Furthermore, it allows pre-symptomatic diagnosis in young family members and definitive screening in potential living kidney donors from PKD families before donation.
Identifying the specific mutation also provides useful information about likely disease course. Consequently, knowing whether a patient carries a PKD1 truncating mutation — the highest-risk type — versus a PKD2 mutation influences the urgency of treatment and monitoring.
Blood and Urine Tests
Blood tests including serum creatinine and eGFR measure current kidney function and provide a baseline for monitoring change over time. Urine albumin-to-creatinine ratio detects early protein loss. Furthermore, regular blood pressure monitoring at home and in clinic is essential because high blood pressure is both an early warning sign and a driver of ADPKD progression. Moreover, urine testing helps detect blood and urinary tract infections, both of which need prompt attention in PKD.
Treatment of Polycystic Kidney Disease
Blood Pressure Control
Tight blood pressure control is the foundation of ADPKD management. ACE inhibitors and ARBs are the preferred blood pressure medications because they reduce pressure both within the kidney’s filtering units and in the wider circulation. Consequently, the landmark HALT-PKD trial showed that targeting low blood pressure in young ADPKD patients significantly slowed total kidney volume growth and reduced strain on the heart.
Tolvaptan — Targeted Therapy for ADPKD
Tolvaptan is the first disease-modifying therapy approved specifically for ADPKD. It blocks a receptor called the vasopressin V2 receptor, reducing cyclic AMP levels within kidney tubular cells — one of the key signals driving cyst growth. The TEMPO 3:4 and REPRISE clinical trials showed that tolvaptan significantly slows total kidney volume growth and preserves eGFR compared with placebo in patients with rapidly progressing ADPKD.
Tolvaptan suits adults with ADPKD who are at risk of rapid progression. However, it carries a risk of serious liver toxicity in a small number of patients. Consequently, liver function monitoring is essential throughout treatment and patients must receive thorough counselling about this risk before starting.
Furthermore, tolvaptan causes a strong increase in urine output. This requires patients to drink very large amounts of fluid throughout treatment. Therefore, patients must be able to manage this requirement in their daily life before tolvaptan is prescribed.
Managing Pain, Infections, and Cyst Complications
Pain management in PKD requires care. Paracetamol is the preferred painkiller because NSAIDs carry risks for patients with reduced kidney function. For severe cyst-related pain, moreover, procedures including cyst aspiration or laparoscopic cyst removal may provide relief when simple measures fail.
Urinary tract infections in PKD need prompt antibiotic treatment. Without treatment, infections can spread into cysts and become very difficult to clear. Furthermore, cyst infections require prolonged courses of antibiotics that can penetrate the cyst wall — such as fluoroquinolones. Consequently, a suspected cyst infection requires specialist input and longer treatment than a simple urinary infection.
Kidney stones occur more often in PKD than in the general population. Stone management is more complex in the context of greatly enlarged kidneys with altered anatomy. Therefore, specialist urological input is important for stone treatment in advanced PKD.
Renal Replacement Therapy and Transplantation
For patients reaching kidney failure, kidney transplantation is the best form of renal replacement therapy. PKD patients generally have very good outcomes after transplantation. Furthermore, the native polycystic kidneys are typically removed at the time of transplantation if they are causing symptoms, infection, or significant displacement of other organs. Consequently, removal of the original kidneys significantly improves abdominal comfort and quality of life.
Haemodialysis and peritoneal dialysis are both options when transplantation is not immediately possible. However, peritoneal dialysis can be technically difficult in patients with greatly enlarged polycystic kidneys and livers that reduce available space in the abdomen. Therefore, haemodialysis is the more commonly chosen option in advanced ADPKD with significant organ enlargement.
Living Well With Polycystic Kidney Disease
Lifestyle Modifications
Several lifestyle changes have real evidence for slowing PKD progression. High water intake — targeting a daily urine output of two to three litres — reduces vasopressin release. This consequently lowers cyclic AMP-driven cyst growth. This simple change is low cost, safe, and supported by physiological evidence. Furthermore, it directly mirrors one of the ways tolvaptan slows cyst growth.
Dietary changes include moderate protein restriction, reduced sodium intake to lower blood pressure and fluid retention, and limiting caffeine — which raises cyclic AMP levels in tubular cells and may encourage cyst growth. Moreover, maintaining a healthy body weight reduces the cardiovascular burden that affects PKD patients more than the general population. Regular aerobic exercise supports blood pressure control, heart health, and mental wellbeing throughout all stages of the disease.
Genetic Counselling and Family Screening
Because ADPKD is an autosomal dominant condition, first-degree relatives of affected individuals have a 50% chance of carrying the mutation. Consequently, family screening — using renal ultrasound or genetic testing — is recommended for all first-degree relatives of a confirmed ADPKD patient. Furthermore, genetic counselling helps families understand how the condition is inherited, what screening results mean, and how to make informed decisions about family planning.
Prenatal genetic testing and preimplantation genetic diagnosis are available for families with confirmed PKD mutations who wish to consider their options before or during pregnancy. These choices need careful discussion with specialist genetic counselling and reproductive medicine teams. Moreover, the personal and ethical dimensions of these decisions deserve thoughtful and unhurried consideration in a supportive clinical setting.
Monitoring and Long-Term Follow-Up
Regular long-term follow-up with a specialist kidney team is essential for all patients with PKD. Annual or twice-yearly eGFR monitoring tracks the rate of kidney function decline. Regular imaging — typically renal ultrasound or MRI — monitors total kidney volume and detects complications such as cyst bleeding, infection, or concerns about cancer.
Furthermore, blood pressure monitoring, heart and blood vessel risk assessment, and screening for non-kidney complications form essential parts of comprehensive PKD care. Screening for brain aneurysms is specifically recommended for ADPKD patients with a family history of aneurysm rupture or brain bleeding. Consequently, long-term monitoring addresses the whole-body nature of PKD rather than focusing on kidney function alone.
When to Seek Urgent Medical Help
Seek emergency medical care immediately if you experience sudden and severe flank pain — which may indicate cyst bleeding, stone passage, or cyst infection. Furthermore, the sudden onset of the worst headache of your life in a person with PKD requires immediate emergency assessment to exclude bleeding around the brain from a ruptured aneurysm.
High fever with flank pain or urinary symptoms suggests a cyst infection needing urgent antibiotic treatment. Moreover, heavy or prolonged blood in the urine, or blood in the urine with clots, needs prompt medical review. Consequently, any sudden change in symptoms in a person with known PKD deserves urgent medical assessment rather than watchful waiting at home.
Frequently Asked Questions About Polycystic Kidney Disease Genetic Cysts Kidneys
1. If my parent has ADPKD, will I definitely develop it?
Not necessarily. Each child of an affected parent has a 50% chance of inheriting the mutated gene. However, if you do not inherit the mutation, you cannot develop ADPKD or pass it to your own children. Furthermore, genetic testing or renal ultrasound screening can determine definitively whether you carry the mutation. Consequently, screening first-degree relatives of affected individuals is strongly recommended to allow early intervention if the mutation is present.
2. Can PKD cysts be removed without surgery?
Individual cysts causing significant pain or local problems can sometimes be treated with ultrasound-guided aspiration and sclerotherapy — a minimally invasive procedure. However, this does not stop the growth of remaining cysts. Furthermore, new cysts continue to form regardless of treatment to individual cysts. Consequently, cyst removal is generally reserved for specific complications rather than used as a general strategy for disease control.
3. Does polycystic kidney disease always lead to kidney failure?
Not always, and not necessarily within a normal lifespan. Many people with PKD2 mutations maintain adequate kidney function into their seventies or eighties. Furthermore, with modern blood pressure control, tolvaptan therapy in eligible patients, and lifestyle changes, the rate of progression can be significantly slowed. Consequently, a diagnosis of PKD does not inevitably mean kidney failure — particularly for those diagnosed early and managed well.
4. Is there a cure for polycystic kidney disease?
There is currently no cure for PKD. Tolvaptan is the only approved disease-modifying therapy and slows rather than stops cyst growth. However, active research continues into several promising targets including mTOR inhibitors, CDK inhibitors, and gene therapy. Furthermore, clinical trials keep testing new agents targeting the cyclic AMP and mTOR pathways. Consequently, the treatment options for PKD are developing rapidly and the long-term outlook for future patients continues to improve.
5. Can people with PKD donate a kidney to a family member?
People with confirmed PKD are not eligible to donate a kidney because the condition affects both kidneys. However, family members who wish to donate must have thorough imaging and ideally genetic testing to confirm they have not inherited the mutation before donation proceeds. Furthermore, young relatives may not yet show cysts on imaging despite carrying the mutation. Consequently, genetic testing rather than imaging alone is the most reliable way to screen potential donors from PKD families.
References
- PCOS is an endocrine and metabolic disorder characterized by insulin resistance
- To promote kidney health, experts recommend focusing on whole, nutrient-rich foods.
- Obesity stands as the strongest modifiable risk factor for endometrial cancer
- Echinococcosis is caused by tapeworms from the genus Echinococcus
Disclaimer
This article is for informational purposes only and does not constitute medical advice. It is not a substitute for professional diagnosis, treatment, or guidance from a licensed healthcare provider. If you have symptoms of polycystic kidney disease or any other medical condition, please consult a qualified doctor promptly. Always follow the advice of your healthcare team for your individual health needs.
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.