
Polycythemia Vera: Too Many Red Blood Cells and Clotting Risk
The human body maintains exquisitely precise control over the number of blood cells circulating at any given time. The bone marrow produces exactly as many red blood cells as the body needs — responding to oxygen levels, hormone signals, and the body’s physiological demands with remarkable accuracy. However, in polycythemia vera, this finely tuned system breaks down. The bone marrow produces far too many red blood cells — regardless of what the body actually needs.
Polycythemia vera is a chronic blood cancer in which a genetic mutation drives the uncontrolled overproduction of red blood cells. The blood becomes abnormally thick and viscous — like syrup rather than fluid. It flows sluggishly through blood vessels, clots far more easily than normal, and generates a dangerous and progressive risk of heart attack, stroke, deep vein thrombosis, and pulmonary embolism.
Polycythemia vera red blood cells clotting risk is the central clinical challenge of this condition. Furthermore, PV — as it is commonly abbreviated — progresses slowly and often causes only vague symptoms for years before a serious thrombotic event brings it to medical attention. Consequently, understanding the disease mechanism, recognising its early warning signs, and pursuing treatment that genuinely reduces clotting risk is essential for every person affected by this under-recognised but dangerous condition.
Quick Answer
Polycythemia vera is a chronic bone marrow cancer driven by a JAK2 gene mutation. It overproduces red blood cells — and often platelets and white cells — making the blood dangerously thick. This increases the risk of serious blood clots including stroke and pulmonary embolism. Treatment with phlebotomy, aspirin, and cytoreductive drugs reduces this risk significantly.
What Is Polycythemia Vera?
A Myeloproliferative Neoplasm
Polycythemia vera belongs to a family of related blood cancers called myeloproliferative neoplasms — abbreviated MPN. This group also includes essential thrombocythaemia — characterised by excess platelets — and primary myelofibrosis — characterised by progressive marrow scarring. All three conditions share a common biological driver — the acquisition of a somatic mutation in haematopoietic stem cells that activates a signalling pathway controlling blood cell production.
In polycythemia vera, this overactivated signalling pathway drives excessive proliferation of all three blood cell lines — red cells, white cells, and platelets — but red cell overproduction dominates. The term polycythemia literally means too many blood cells. The term vera — Latin for true — distinguishes the condition from secondary polycythaemia — the reactive increase in red cells that occurs in response to chronic low oxygen states such as altitude exposure or lung disease. Consequently, the name itself encapsulates the fundamental problem — a primary, intrinsic overproduction of blood cells by a genetically abnormal marrow clone.
How Excess Red Blood Cells Harm the Body
In a healthy adult, the haematocrit — the percentage of blood volume occupied by red cells — ranges from approximately 40 to 52% in men and 36 to 48% in women. In polycythemia vera, the haematocrit rises well above these levels — often reaching 55 to 65% without treatment. At these levels, blood viscosity — its thickness and resistance to flow — increases dramatically.
Thick blood flows more slowly through vessels. It generates higher shear stress on vessel walls. It activates platelets and clotting factors more readily. Furthermore, the sluggish flow creates areas of stasis — where blood stops moving entirely — in veins and small vessels. Stasis is one of the three classic drivers of blood clot formation — alongside vessel wall damage and clotting factor activation. Consequently, polycythemia vera creates a persistently pro-thrombotic environment that dramatically elevates the risk of clotting events throughout the vascular system.
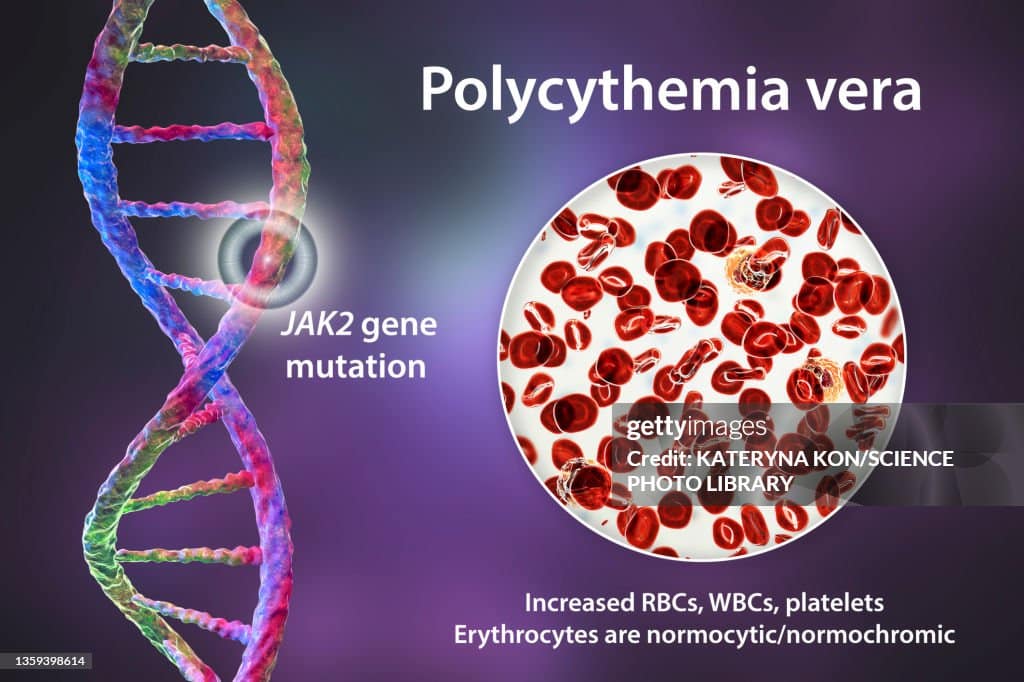
The JAK2 Mutation — The Driver of Polycythemia Vera
What JAK2 Does Normally
JAK2 — Janus kinase 2 — is an enzyme that plays a central role in transmitting signals from erythropoietin — the hormone that stimulates red blood cell production — into haematopoietic stem cells. When erythropoietin binds to its receptor on stem cell surfaces, JAK2 activates and triggers a cascade of intracellular signals that instruct the cell to divide and produce red blood cells.
Under normal conditions, JAK2 activates only when erythropoietin binding occurs — providing a tightly regulated, on-demand signal for red cell production. When erythropoietin levels fall — because the body already has enough oxygen-carrying red cells — JAK2 activity ceases and red cell production stops. This feedback loop keeps haematocrit within its narrow physiological range with remarkable precision.
The V617F Mutation
In polycythemia vera, a specific point mutation in the JAK2 gene — called JAK2 V617F — permanently activates the JAK2 enzyme regardless of erythropoietin levels. The mutant JAK2 enzyme behaves as if erythropoietin is permanently present and permanently instructing the cell to produce red blood cells. Consequently, the stem cells carrying this mutation produce red cells continuously and uncontrollably — completely uncoupled from the body’s actual oxygen needs.
The JAK2 V617F mutation is found in roughly 95% of polycythemia vera patients. The remaining 5% carry alternative mutations in exon 12 of the JAK2 gene that produce the same constitutive activation effect. Furthermore, JAK2 mutations are also found in essential thrombocythaemia and primary myelofibrosis — though with different frequencies and clinical consequences. Consequently, JAK2 V617F testing is now the cornerstone of PV diagnosis — providing both diagnostic confirmation and a target for molecular therapies. For context on how somatic mutations drive related bone marrow disorders, see our article on myelodysplastic syndrome — the pre-leukemia condition explained.
Symptoms of Polycythemia Vera
Vascular and Circulatory Symptoms
Polycythemia vera red blood cells clotting risk produces a distinctive symptom pattern. Headache is among the most common early symptoms — arising from increased blood viscosity and slowed cerebral blood flow. The headaches are often persistent, dull, and poorly responsive to standard analgesics. Furthermore, dizziness, visual disturbances — including blurring and transient vision loss — and difficulty concentrating all reflect inadequate cerebral perfusion from thickened blood.
Facial redness — called plethora — is a characteristic visible sign. The face, particularly the cheeks and nose, takes on a deep red or purplish hue from the engorged superficial blood vessels filled with excess red cells. The palms and fingertips may also appear reddened. Moreover, ringing in the ears — tinnitus — and a feeling of fullness or pressure in the head reflect the increased blood volume and viscosity affecting cranial circulation. Consequently, the cluster of headache, dizziness, visual symptoms, and facial redness in an older adult should prompt immediate blood count investigation.
Itching, Splenomegaly, and Gout
Aquagenic pruritus — intense itching triggered by contact with water, particularly warm or hot water — is one of the most diagnostically distinctive symptoms of polycythemia vera. It occurs in roughly 40% of patients and can be severely disabling. The itching typically begins within minutes of bathing and subsides over thirty to sixty minutes. Furthermore, it has no visible skin lesion — the skin appears normal despite the intense itch. The mechanism involves histamine release from activated mast cells and basophils in response to the abnormal blood composition.
Splenomegaly — enlargement of the spleen — develops as the spleen expands its role in processing the excess red cells circulated by the overactive marrow. Patients notice a feeling of fullness, early satiety after small meals, and left upper abdominal discomfort or pain. Moreover, gout — caused by elevated uric acid from the accelerated turnover of excess blood cells — develops in a proportion of PV patients. Elevated uric acid levels reflect the high cell turnover rate accompanying any myeloproliferative condition. For context on how autoimmune and systemic conditions produce multi-system effects, see our article on lupus nephritis — when lupus attacks the kidneys.
Thrombotic Events
Blood clots represent the most serious and life-threatening complication of polycythemia vera. Arterial thrombosis — clotting in arteries — causes stroke, transient ischaemic attack, myocardial infarction, and peripheral arterial occlusion. Venous thrombosis — clotting in veins — causes deep vein thrombosis, pulmonary embolism, and — characteristically for PV — thrombosis in unusual sites including the hepatic veins causing Budd-Chiari syndrome, the portal vein, mesenteric veins, and cerebral venous sinuses.
Thrombosis in unusual venous sites — particularly the hepatic veins or portal vein — in a young or middle-aged adult should always prompt JAK2 mutation testing to exclude polycythemia vera as the underlying cause. Furthermore, up to 30% of PV patients have already experienced a thrombotic event before their diagnosis is established — reflecting the delayed recognition problem inherent in a condition whose early symptoms are non-specific. Consequently, thrombosis prevention is the primary treatment goal throughout the management of polycythemia vera.
How Doctors Diagnose Polycythemia Vera
Blood Count Findings
Diagnosing polycythemia vera red blood cells clotting risk begins with a full blood count. The characteristic findings are an elevated haemoglobin — above 165 grams per litre in men or 160 grams per litre in women — or an elevated haematocrit — above 49% in men or 48% in women — combined with elevated red cell mass. Furthermore, the white blood cell count is elevated in roughly 60% of patients at diagnosis. Platelet count is elevated in roughly 50% of patients. Consequently, the combination of elevated counts across multiple cell lines — called panmyelosis — strongly suggests a myeloproliferative process rather than a secondary reactive polycythaemia.
The blood film shows increased numbers of morphologically normal-appearing red cells — distinguishing PV from myelodysplastic syndrome where dysplastic red cell forms are prominent. Furthermore, basophilia — elevated basophil count — is a useful supportive finding because reactive polycythaemia does not cause basophilia while PV frequently does. Consequently, these blood count patterns together provide strong pre-test probability for PV before molecular testing confirms the diagnosis.
JAK2 Mutation Testing and WHO Criteria
JAK2 V617F mutation testing on peripheral blood — using polymerase chain reaction — provides diagnostic confirmation in approximately 95% of cases. A positive result in the context of elevated haemoglobin and haematocrit essentially confirms PV without the need for bone marrow biopsy in most straightforward cases.
The 2016 WHO diagnostic criteria for polycythemia vera require meeting three major criteria or two major criteria plus one minor criterion. The major criteria are elevated haemoglobin or haematocrit as defined above, the presence of JAK2 V617F or JAK2 exon 12 mutation, and bone marrow biopsy showing hypercellularity with panmyelosis — proliferation of all three cell lineages. The minor criterion is a subnormal erythropoietin level. Consequently, erythropoietin measurement plays a supporting diagnostic role — PV typically produces low erythropoietin levels because the feedback loop driving erythropoietin release is suppressed by the constitutively high red cell mass.
Bone Marrow Biopsy
Bone marrow biopsy is not required in all PV cases — particularly when JAK2 mutation positivity and blood count criteria clearly confirm the diagnosis. However, biopsy provides important additional information. It confirms the characteristic hypercellular, panmyelotic marrow pattern. Furthermore, it assesses for fibrosis — the degree of reticulin fibrosis in the marrow predicts the risk of progression to post-PV myelofibrosis. Consequently, baseline biopsy is recommended for most newly diagnosed patients to provide a morphological reference point for monitoring disease evolution over subsequent years.
Treatment of Polycythemia Vera
Phlebotomy — The First-Line Treatment
Phlebotomy — the therapeutic removal of blood — is the cornerstone of initial PV treatment. Removing 400 to 500 millilitres of blood at each session rapidly reduces haematocrit toward the target range. The target haematocrit is below 45% — a level demonstrated by the CYTO-PV clinical trial to significantly reduce the risk of major thrombotic events and cardiovascular death compared with a less strict target of 45 to 50%.
Phlebotomy sessions begin as frequently as every two to three days in newly diagnosed patients with very high haematocrit — gradually reducing to once every four to eight weeks once the target is achieved and maintained. Furthermore, phlebotomy depletes iron stores over time — limiting the marrow’s capacity to produce excess red cells and consequently extending the interval between sessions in many patients. Phlebotomy does not reduce platelet counts or white cell counts — meaning cytoreductive drug therapy is needed for patients with elevated counts from these lineages contributing to thrombotic risk.
Low-Dose Aspirin
Low-dose aspirin — typically 75 to 100 milligrams daily — is recommended for all PV patients without contraindication. It inhibits platelet aggregation and reduces the risk of arterial thrombotic events including stroke and myocardial infarction. The ECLAP study demonstrated that low-dose aspirin significantly reduced the risk of combined cardiovascular events — including fatal and non-fatal thrombosis — without a significant increase in major bleeding complications in PV patients. Consequently, aspirin is universally incorporated into PV treatment regimens alongside haematocrit control.
Cytoreductive Therapy
Cytoreductive therapy — using drugs to reduce bone marrow blood cell production — is indicated for high-risk PV patients. High-risk features include age over 60, a prior history of thrombotic events, or very high platelet counts. Furthermore, patients who require excessively frequent phlebotomy to maintain haematocrit target or who have poorly controlled symptoms also benefit from cytoreductive treatment.
Hydroxyurea — also called hydroxycarbamide — is the standard first-line cytoreductive agent. It inhibits DNA synthesis in rapidly dividing cells — suppressing the overactive myeloproliferative clone. Hydroxyurea reduces haematocrit, white cell count, and platelet count simultaneously. Furthermore, it reduces the frequency of phlebotomy required and improves symptom control including splenomegaly and pruritus. Oral administration once or twice daily makes it practical for long-term outpatient use. Consequently, hydroxyurea achieves good haematological control in the majority of PV patients who require cytoreduction.
Managing Hydroxyurea Resistance and Intolerance
Roughly 20 to 25% of PV patients become resistant or intolerant to hydroxyurea over time. Resistance manifests as failure to maintain target haematocrit or rising blood counts despite adequate hydroxyurea dosing. Intolerance occurs through drug-related side effects including skin ulceration, mouth sores, and — rarely — secondary malignancy concern with very prolonged use.
Ruxolitinib — a JAK1 and JAK2 inhibitor — is the most effective second-line cytoreductive treatment for hydroxyurea-resistant or intolerant PV. The RESPONSE clinical trial demonstrated that ruxolitinib achieved haematocrit control and spleen volume reduction significantly more effectively than standard alternative therapies. Furthermore, ruxolitinib dramatically reduces aquagenic pruritus and constitutional symptoms — producing meaningful quality of life improvements beyond haematological control. Ropeginterferon alfa-2b — a long-acting interferon preparation — is an alternative cytoreductive option with the unique advantage of reducing the JAK2 allele burden — the proportion of cells carrying the mutant JAK2 — and producing molecular remissions in some patients. Consequently, molecular response offers the prospect of deeper and potentially more durable disease control than haematological response alone.
Disease Progression and Long-Term Monitoring
Transformation to Myelofibrosis and Leukaemia
Polycythemia vera is a progressive disease. Over ten to twenty years, roughly 15 to 20% of PV patients develop post-PV myelofibrosis — a transformation in which progressive scarring replaces the overactive bone marrow, destroys normal blood production, and causes massive spleen enlargement. Furthermore, roughly 5 to 10% of PV patients develop acute myeloid leukaemia — a transformation carrying a very poor prognosis given the advanced age and prior cytotoxic treatment of most PV patients at the time of transformation.
The risk of both transformations increases with the duration of PV, the degree of splenomegaly, the JAK2 allele burden, and prior exposure to certain cytoreductive agents. Consequently, regular monitoring — including annual or biennial bone marrow biopsy in patients showing clinical signs of progression, serial blood counts, and spleen size assessment — allows early detection of transformation and timely escalation of treatment. For context on how related bone marrow conditions are monitored and managed, see our article on aplastic anemia — when bone marrow stops making blood cells.
Cardiovascular Risk Management
Cardiovascular risk management is as important as haematological control in PV. High blood pressure accelerates vascular damage and multiplies thrombotic risk in patients whose blood is already thick and prone to clotting. Consequently, blood pressure control — targeting below 130 over 80 millimetres of mercury — is a treatment priority in all PV patients with hypertension.
Diabetes, high cholesterol, smoking, and obesity all independently elevate thrombotic risk and must be actively managed alongside PV-specific treatment. Furthermore, regular physical activity — within limits set by individual symptoms and disease status — supports cardiovascular health and reduces venous stasis risk. For context on how kidney function interacts with cardiovascular risk management in chronic conditions, see our article on chronic kidney disease — stages, symptoms, and how to slow the decline.
When to Seek Urgent Medical Help
Seek emergency care immediately if any of the following develop in a person with known or suspected polycythemia vera. Sudden weakness, speech difficulty, facial drooping, or vision loss may indicate stroke. Chest pain or breathlessness suggests pulmonary embolism or myocardial infarction. Severe abdominal pain with fever may indicate mesenteric or portal vein thrombosis. Furthermore, sudden severe headache — the worst headache of a person’s life — requires emergency assessment to exclude cerebral venous sinus thrombosis.
Consequently, patients and families must have a clear emergency action plan and seek immediate hospital assessment without delay when these warning signs appear. Every minute of treatment delay in acute thrombosis increases the extent of irreversible organ damage.
Frequently Asked Questions
1. Is polycythemia vera curable?
Polycythemia vera is not currently curable with standard treatments. Phlebotomy, aspirin, and cytoreductive drugs control the disease effectively — reducing clotting risk and managing symptoms — but do not eliminate the underlying JAK2-mutant clone. Allogeneic stem cell transplantation offers the only realistic chance of cure but carries substantial risks that limit its use to younger patients with aggressive or transformed disease. Furthermore, ropeginterferon alfa-2b achieves deep molecular responses in some patients — reducing the mutant clone significantly — but durable cure even with this agent remains unproven. Consequently, the goal of current treatment is long-term disease control and complication prevention rather than cure.
2. How serious is the clotting risk in polycythemia vera?
The clotting risk in PV is very significant. Thrombotic events — including stroke, myocardial infarction, deep vein thrombosis, and pulmonary embolism — represent the leading cause of morbidity and mortality in PV patients. Without treatment, the annual thrombotic event rate approaches 5% per year in high-risk patients. Furthermore, up to 30% of PV patients have already suffered a thrombotic event before their diagnosis. Consequently, maintaining haematocrit below 45% and taking low-dose aspirin are critical interventions that all patients should adhere to consistently throughout their care.
3. Can polycythemia vera be confused with other conditions?
Yes. PV is frequently confused with secondary polycythaemia — the reactive increase in red cells caused by chronic hypoxia from lung disease, sleep apnoea, or high-altitude living. Secondary polycythaemia does not carry the intrinsic clotting risk of PV and does not require the same treatment. Furthermore, PV is sometimes confused with essential thrombocythaemia — another JAK2-associated myeloproliferative neoplasm — particularly when thrombocytosis is a dominant finding. Consequently, JAK2 testing and erythropoietin measurement are essential to accurately classify the myeloproliferative disorder and direct the correct treatment.
4. What triggers the JAK2 mutation in polycythemia vera?
The precise trigger for the JAK2 V617F mutation in most PV patients remains unknown. The mutation arises as a spontaneous somatic error — a random DNA copying mistake — in a haematopoietic stem cell during normal cell division. It is not inherited in most cases and does not result from a known external exposure in the majority of patients. Furthermore, advancing age increases the probability of acquiring such mutations simply through the cumulative number of cell divisions occurring over a lifetime. Consequently, most PV patients have no identifiable cause for their JAK2 mutation beyond the biological reality of ageing bone marrow.
5. Does polycythemia vera affect life expectancy?
With modern treatment, most PV patients achieve near-normal life expectancy — particularly those who maintain good haematocrit control and cardiovascular risk management. The main threats to life expectancy are thrombotic events — which appropriate treatment dramatically reduces — and long-term disease transformation to myelofibrosis or leukaemia. Furthermore, patients diagnosed before the age of 60 — when they have the longest potential disease duration — benefit most from consistent long-term management and regular specialist monitoring. Consequently, PV should not be viewed as a rapidly fatal disease — but rather as a chronic condition requiring lifelong attention and engagement with specialist haematological care.
References
- Competitive power generation costs make investment in renewables highly attractive as countries target economic recovery from COVID-19, new IRENA report finds.
- India is set to significantly enhance its solar photovoltaic (PV) module manufacturing capacity, increasing from 80 gigawatts (GW) to 125 GW by 2030.
- Aloe vera, scientifically known as Aloe barbadensis Miller, isn’t just another houseplant.
- Scientists have categorized dark comets into two distinct groups: smaller inner dark comets and larger outer dark comets.
Disclaimer
This article adapts publicly available information from WHO’s Blood Safety and Availability page. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform and not a healthcare provider.
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.