
Myelodysplastic Syndrome: The Pre-Leukemia Condition Explained
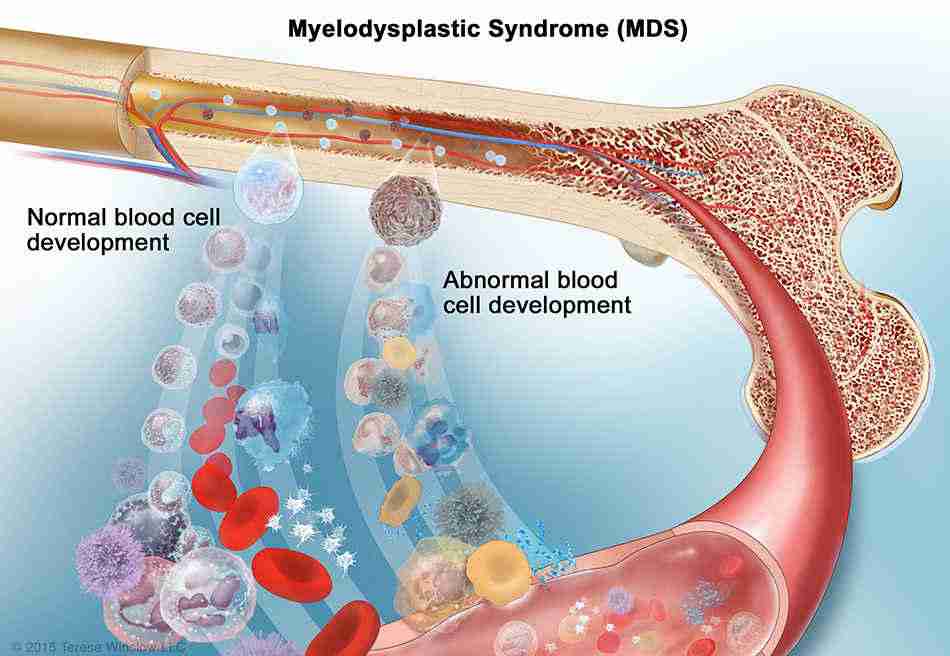
Blood is produced quietly and continuously inside the bone marrow. Most people never think about this process — it simply happens, reliably, every second of every day. However, for some people, something goes wrong at the most fundamental level. The blood cells that emerge from the marrow are abnormal — misshapen, dysfunctional, and short-lived. The body’s blood supply slowly deteriorates, and the risk of a more dangerous disease begins to grow.
Myelodysplastic syndrome — commonly called MDS — is a group of bone marrow disorders in which the marrow produces blood cells that are abnormal in appearance and function. These dysplastic cells — meaning abnormally formed cells — fail to mature properly, die prematurely, and accumulate genetic mutations over time. As a result, blood cell counts fall, symptoms of bone marrow failure develop, and a proportion of patients progress to acute myeloid leukaemia — one of the most aggressive blood cancers.
Myelodysplastic syndrome pre-leukemia undetected diagnosis is the central problem this article addresses. MDS is notoriously difficult to detect in its early stages because its initial symptoms — fatigue, mild anaemia, and occasional infections — are non-specific and easily attributed to ageing or other common conditions. Furthermore, it predominantly affects adults over 65 — a population where such symptoms are frequently dismissed as expected accompaniments of older age. Consequently, understanding MDS clearly — what it is, who develops it, and how it is diagnosed and treated — is essential for patients, families, and healthcare providers alike.
Quick Answer
Myelodysplastic syndrome is a group of bone marrow disorders in which abnormal blood cells fail to mature properly, leading to low blood cell counts and risk of progression to acute myeloid leukaemia. It predominantly affects older adults and often goes undetected until blood tests reveal anaemia or other abnormalities. Treatment ranges from supportive care to stem cell transplantation.
What Is Myelodysplastic Syndrome?
Dysplasia and Bone Marrow Failure
Myelodysplastic syndrome pre-leukemia undetected diagnosis begins with understanding what MDS actually does to the bone marrow. The word myelodysplastic combines three Greek roots — myelo meaning marrow, dys meaning abnormal, and plastic meaning formation. Together, the term describes a condition of abnormal cell formation in the bone marrow.
In healthy bone marrow, haematopoietic stem cells — the master blood-producing cells — divide and mature through a precise sequence of developmental stages. Each stage produces increasingly specialised cells that ultimately become functional red blood cells, white blood cells, or platelets. This process, called haematopoiesis, operates with extraordinary reliability throughout life.
In MDS, genetic mutations accumulate in haematopoietic stem cells and disrupt this maturation process. The abnormal stem cells still divide — sometimes excessively — but they produce cells that fail to mature correctly. These immature, dysplastic cells accumulate in the marrow and spill into the bloodstream. Furthermore, they die prematurely — a process called ineffective haematopoiesis — leaving the body with fewer functional blood cells than it needs. Consequently, the bone marrow appears hypercellular — packed with cells — yet the blood paradoxically shows low counts because most cells produced are dysfunctional.
The Relationship to Acute Myeloid Leukaemia
MDS sits in a biological continuum between normal bone marrow and acute myeloid leukaemia — abbreviated AML. As genetic mutations accumulate in dysplastic stem cells over time, a clone of cells may acquire additional mutations that drive uncontrolled proliferation. When the proportion of immature blast cells — the precursors to all blood cells — exceeds 20% of bone marrow cells, the diagnosis shifts from MDS to AML.
This progression does not occur in all MDS patients. Roughly 30% of MDS cases transform to AML over their natural history. However, the risk of transformation varies enormously — from less than 5% in low-risk MDS subtypes to more than 40% in high-risk subtypes within two years. Furthermore, even patients who do not transform to AML face significant morbidity and mortality from the complications of bone marrow failure itself — infection, bleeding, and transfusion dependence. For context on how bone marrow failure affects blood cell production across related conditions, see our article on aplastic anemia — when bone marrow stops making blood cells.
What Causes Myelodysplastic Syndrome?
Acquired Genetic Mutations
The fundamental cause of MDS is the acquisition of genetic mutations in haematopoietic stem cells. These mutations disrupt the genes controlling cell growth, differentiation, and programmed cell death. Over time, the mutant clone expands and progressively displaces normal stem cells from the bone marrow.
Somatic mutations — meaning mutations that arise in individual body cells during life rather than being inherited — drive most MDS cases. Genes most frequently mutated in MDS include SF3B1 — involved in RNA splicing — TET2 and DNMT3A — involved in gene expression regulation — TP53 — the major tumour suppressor gene — and genes controlling the RAS signalling pathway. Furthermore, chromosomal abnormalities — deletions, duplications, and rearrangements of entire chromosome segments — are identified in roughly 50% of MDS cases. The deletion of chromosome 5q — del(5q) — is the most common and clinically important chromosomal abnormality in MDS.
Prior Treatment and Environmental Exposure
Prior chemotherapy or radiation therapy for another cancer represents the most clearly established environmental cause of MDS. This is called therapy-related MDS — abbreviated t-MDS — and typically develops five to ten years after the prior treatment. Alkylating agents — a class of chemotherapy drugs — and topoisomerase II inhibitors both carry well-established MDS risk. Furthermore, radiation therapy to the pelvis or spine — sites of active bone marrow — directly damages haematopoietic stem cells and increases mutation accumulation.
Benzene — an industrial chemical solvent — is the most important non-iatrogenic environmental trigger. Occupational benzene exposure in industries including petroleum refining, rubber manufacturing, and shoe production is associated with significantly elevated MDS risk. Moreover, heavy cigarette smoking — which delivers benzene and other haematopoietic toxins — increases MDS risk in a dose-dependent manner. Consequently, occupational and lifestyle history is an important component of MDS evaluation in every newly diagnosed patient.
Age and Clonal Haematopoiesis
MDS is predominantly a disease of older adults. The median age at diagnosis is approximately 70 years. This age predilection reflects the cumulative acquisition of somatic mutations in haematopoietic stem cells over a lifetime. The more times a stem cell divides — and stem cells divide billions of times over decades — the more opportunities exist for replication errors to introduce pathogenic mutations.
An important concept related to MDS is clonal haematopoiesis of indeterminate potential — abbreviated CHIP. This refers to the presence of MDS-associated somatic mutations in blood cells without the blood count abnormalities needed to diagnose MDS. CHIP is extremely common in older adults — detectable in roughly 10 to 20% of people over 70. Furthermore, CHIP carriers face an elevated risk of eventually progressing to MDS or AML — though the annual risk of progression remains low at roughly 0.5 to 1% per year. Consequently, CHIP represents a pre-MDS state that requires monitoring rather than immediate treatment.
Symptoms of Myelodysplastic Syndrome
Anaemia and Fatigue
The symptoms of myelodysplastic syndrome pre-leukemia undetected diagnosis largely reflect the consequences of reduced blood cell production. Anaemia — low red blood cell count — is the most common finding at presentation. It causes fatigue, weakness, pallor, and shortness of breath on exertion. Furthermore, the anaemia of MDS tends to be macrocytic — meaning the individual red blood cells are abnormally large, though few in number — a pattern visible on blood film examination that provides an important diagnostic clue.
The fatigue of MDS is often profound and disproportionate to the degree of anaemia — reflecting not only low haemoglobin but also the systemic effect of cytokine dysregulation from the disordered marrow. Many patients describe months or years of gradually worsening fatigue before diagnosis — during which they and their doctors attributed the symptom to ageing, stress, or other common conditions. Consequently, MDS frequently presents late because its initial symptoms blend imperceptibly into the background of older adult health complaints.
Infections and Bleeding
Low white blood cell count — particularly neutropenia — impairs infection defence and produces recurrent bacterial and fungal infections. Mouth ulcers, skin infections, urinary tract infections, and pneumonia recur more frequently than expected. Furthermore, infections that a healthy immune system would clear rapidly become prolonged and difficult to treat in neutropenic MDS patients.
Low platelet count — thrombocytopenia — produces the bleeding manifestations of MDS. Easy bruising, petechiae — tiny skin haemorrhages — prolonged bleeding from minor cuts, frequent nosebleeds, and bleeding gums all reflect impaired platelet-mediated clotting. Spontaneous internal bleeding — including gastrointestinal bleeding and intracranial haemorrhage — occurs when platelet counts fall to critically low levels. Moreover, the combination of recurrent infections and unusual bleeding in an older adult should always prompt blood count investigation to exclude MDS. For context on how infection and bleeding complicate bone marrow failure conditions, see our article on aplastic anemia — when bone marrow stops making blood cells.
Splenomegaly and Other Features
Some MDS patients develop splenomegaly — enlargement of the spleen — as the spleen expands its extramedullary haematopoiesis — producing blood cells outside the marrow — to compensate for failing marrow function. Patients notice a feeling of fullness or discomfort in the left upper abdomen. Furthermore, an enlarged spleen accelerates platelet and red cell destruction — worsening the cytopenias that the marrow can no longer correct.
Systemic symptoms including fever, night sweats, and unintentional weight loss occur in some MDS patients — particularly those with higher-risk disease. These constitutional symptoms reflect the systemic inflammatory state generated by the disordered marrow and the cytokine dysregulation that accompanies advanced clonal haematopoietic disease. Consequently, their presence indicates more aggressive disease biology and a higher risk of progression to AML.
How Doctors Diagnose Myelodysplastic Syndrome
Blood Count and Film Analysis
Diagnosing myelodysplastic syndrome pre-leukemia undetected diagnosis begins with a full blood count showing cytopenia — low counts of one or more blood cell types. Anaemia is present in roughly 90% of MDS patients at diagnosis. Furthermore, neutropenia and thrombocytopenia are present in varying combinations depending on the MDS subtype.
The blood film provides critical morphological information. Dysplastic red cells — including abnormally large cells called macrocytes, oval-shaped macrocytes called macro-ovalocytes, and red cells with abnormal nuclear fragments called Howell-Jolly bodies — suggest MDS. Hypogranular or hyposegmented neutrophils — called pseudo-Pelger-Huët cells — indicate dysplastic white cell production. Moreover, giant platelets and abnormal platelet granularity reflect megakaryocyte dysplasia in the marrow. Consequently, an experienced haematologist reviewing the blood film can raise strong diagnostic suspicion for MDS before bone marrow investigation.
Bone Marrow Examination
Bone marrow aspiration and biopsy are mandatory for MDS diagnosis and subtype classification. Aspiration — withdrawing liquid marrow through a needle — provides cells for morphological assessment, flow cytometry — measuring cell surface protein expression — and cytogenetics — chromosome analysis. Biopsy — extracting a core of solid marrow tissue — assesses overall cellularity and architecture.
Morphological examination quantifies the percentage of blast cells — the critical measurement distinguishing MDS subtypes from each other and from AML. Dysplasia affecting 10% or more of cells in any lineage confirms the diagnosis. Furthermore, cytogenetic analysis — conventional karyotyping and fluorescence in situ hybridisation — identifies chromosomal abnormalities including del(5q), monosomy 7, and complex karyotype that critically influence prognosis and treatment selection. Molecular genetic testing — using next-generation sequencing to detect somatic mutations — provides additional prognostic information and identifies mutations that predict response to specific treatments. Consequently, a complete bone marrow evaluation including morphology, cytogenetics, and molecular genetics is essential for all newly diagnosed MDS patients.
Risk Stratification With IPSS-R
The Revised International Prognostic Scoring System — called IPSS-R — combines bone marrow blast percentage, cytogenetic risk category, haemoglobin level, platelet count, and neutrophil count into a composite score that predicts both overall survival and the probability of AML transformation. Scores classify patients into five risk groups — very low, low, intermediate, high, and very high.
IPSS-R risk category directly drives treatment decisions. Low and very low risk patients may be managed with supportive care or low-intensity treatment for years. High and very high risk patients face a median survival measured in months without aggressive treatment and require prompt consideration of high-intensity therapy including stem cell transplantation. Consequently, IPSS-R scoring is the most important clinical tool in MDS management and must be calculated for every newly diagnosed patient.
Treatment of Myelodysplastic Syndrome
Supportive Care
Treatment of myelodysplastic syndrome pre-leukemia undetected diagnosis begins with supportive care — the management of cytopenias and their complications — regardless of disease risk category. Red blood cell transfusions correct symptomatic anaemia and maintain haemoglobin at levels that allow acceptable quality of life. Platelet transfusions prevent serious bleeding when platelet counts fall to dangerous levels. Furthermore, erythropoiesis-stimulating agents — proteins that stimulate red blood cell production — reduce transfusion requirements in a subset of MDS patients with low endogenous erythropoietin levels and lower-risk disease.
Luspatercept — a newer erythroid maturation agent — produces transfusion independence in a significant proportion of patients with ring sideroblast MDS — a subtype characterised by iron-loaded mitochondria in developing red cells — who have failed or are ineligible for erythropoiesis-stimulating agents. Furthermore, granulocyte colony-stimulating factor temporarily boosts neutrophil counts and reduces infection risk during periods of severe neutropenia. Antimicrobial prophylaxis with antibiotics and antifungal agents protects profoundly neutropenic patients from the opportunistic infections that pose the greatest immediate threat to their lives.
Disease-Modifying Treatment
Lenalidomide — an immunomodulatory drug — produces remarkable red cell responses in MDS patients with isolated del(5q) chromosomal abnormality. It achieves transfusion independence in roughly 60 to 70% of del(5q) patients and in many cases produces cytogenetic remission — disappearance of the abnormal chromosome. Consequently, lenalidomide is the standard first-line disease-modifying treatment for del(5q) MDS.
Hypomethylating agents — azacitidine and decitabine — represent the primary disease-modifying treatment for higher-risk MDS patients who are not candidates for stem cell transplantation. These drugs work by reversing abnormal gene silencing — restoring expression of genes that suppress cancer cell growth and promote normal differentiation. Azacitidine improves blood counts, reduces transfusion dependence, delays AML transformation, and — in the pivotal AZA-001 clinical trial — significantly improved overall survival compared with conventional chemotherapy regimens. Furthermore, decitabine achieves comparable response rates and represents a viable alternative particularly in patients with specific molecular profiles. Consequently, hypomethylating agents form the backbone of higher-risk MDS treatment outside the transplant setting.
Stem Cell Transplantation
Allogeneic stem cell transplantation — replacing the patient’s diseased marrow with donor haematopoietic stem cells — is the only potentially curative treatment for MDS. It eliminates the abnormal MDS clone and reconstitutes a healthy donor-derived blood production system. Transplantation achieves long-term disease-free survival in 30 to 50% of MDS patients — varying significantly with patient age, disease risk, donor compatibility, and transplant centre experience.
Transplantation is most appropriate for younger patients — typically under 70 — with intermediate, high, or very high risk MDS who have a suitable donor and adequate organ function to tolerate the procedure. However, reduced-intensity conditioning regimens — using lower doses of chemotherapy and radiation before transplantation than traditional myeloablative approaches — have extended transplant eligibility to older and less fit patients. Furthermore, hypomethylating agent therapy before transplantation — called bridge therapy — reduces blast percentage and improves disease control in higher-risk patients awaiting donor identification. Consequently, early referral to a transplant centre is recommended for all MDS patients who might be transplant candidates, allowing timely donor search and transplant planning.
Living With Myelodysplastic Syndrome
Managing Transfusion Dependence
Many MDS patients become transfusion-dependent — requiring regular red blood cell transfusions to maintain adequate haemoglobin levels and quality of life. Each transfusion delivers iron — contained within haemoglobin — that the body cannot excrete. Over time, iron accumulates in the liver, heart, and endocrine organs — a condition called transfusion-related iron overload. This iron excess causes progressive organ damage — including liver cirrhosis, cardiac arrhythmias, and diabetes. For context on how chronic disease affects multiple organ systems simultaneously, see our article on chronic kidney disease — stages, symptoms, and how to slow the decline.
Iron chelation therapy — using oral agents including deferasirox or injectable desferrioxamine to remove excess iron — reduces organ damage in transfusion-dependent MDS patients with lower-risk disease and longer expected survival. Furthermore, monitoring serum ferritin — a marker of total body iron stores — guides the timing and intensity of chelation therapy. Consequently, iron overload management is an important but often overlooked component of comprehensive MDS care.
Psychological and Quality of Life Support
An MDS diagnosis carries significant psychological weight. The uncertainty of prognosis — not knowing whether the disease will remain stable for years or progress to leukaemia within months — generates chronic anxiety that affects sleep, relationships, and daily functioning. Furthermore, transfusion dependence — requiring regular hospital visits for blood products — disrupts work, travel, and independence in ways that compound the psychological burden.
Psychological support, patient education, and peer support groups significantly improve coping, treatment adherence, and overall quality of life in MDS patients. Moreover, advance care planning — discussing values, preferences, and goals of care with the medical team and family — helps patients maintain a sense of control and ensures treatment decisions align with personal priorities throughout the disease course. Consequently, psychosocial support is as integral to MDS management as any specific drug or procedure.
When to Seek Medical Help
See a doctor promptly if a routine blood test shows unexplained anaemia — particularly if haemoglobin is falling progressively over successive tests. Furthermore, seek medical attention for persistent fatigue that is worsening despite adequate sleep and rest, recurrent infections occurring more frequently or severely than usual, unusual bruising or bleeding from minor injuries, or unexplained weight loss in an older adult.
Consequently, any older adult with unexplained cytopenia — low counts of one or more blood cell types — on a blood test should receive haematology referral and bone marrow investigation to exclude MDS before less likely explanations are pursued.
Frequently Asked Questions
1. Is myelodysplastic syndrome a type of cancer?
MDS sits in a category between benign bone marrow disease and frank blood cancer. Regulatory agencies and cancer registries classify MDS as a haematological malignancy — meaning it shares the fundamental biology of cancer including clonal expansion, genetic instability, and malignant transformation potential. Furthermore, it is treated by oncologists and haematologists using the same framework applied to blood cancers. However, lower-risk MDS behaves more like a chronic bone marrow failure condition than an aggressive cancer — progressing slowly and sometimes remaining stable for years. Consequently, individual prognosis varies enormously across the disease spectrum.
2. How is MDS different from aplastic anemia?
Both conditions cause bone marrow failure and pancytopenia — low counts of all blood cell types. However, their mechanisms differ fundamentally. Aplastic anemia involves immune destruction of stem cells — leaving the marrow hypocellular and empty. MDS involves genetic mutations in stem cells that produce abnormal, dysplastic cells — leaving the marrow typically hypercellular but dysfunctional. Furthermore, MDS carries a risk of AML transformation that aplastic anemia does not. Consequently, bone marrow biopsy morphology is the critical investigation that distinguishes the two conditions and directs appropriate treatment.
3. Can MDS be cured?
For patients who are candidates for allogeneic stem cell transplantation and have a compatible donor, MDS can be cured. Transplantation achieves long-term disease-free survival in 30 to 50% of treated patients. However, for the majority of MDS patients — who are too old or medically unfit for transplantation — treatment controls the disease rather than curing it. Consequently, the realistic goals for most patients are reducing transfusion dependence, preventing or delaying AML transformation, managing complications, and maintaining quality of life for as long as possible.
4. What is the prognosis for myelodysplastic syndrome?
Prognosis varies enormously across the MDS spectrum. Patients with very low risk MDS — as classified by IPSS-R — have a median survival exceeding five years and a low probability of AML transformation. Patients with very high risk MDS have a median survival measured in months without aggressive treatment and a high probability of AML transformation. Furthermore, age, comorbidities, performance status, and access to transplantation all significantly modify individual prognosis. Consequently, IPSS-R risk classification is the essential starting point for any meaningful prognosis discussion.
5. Should all MDS patients receive chemotherapy?
No. Lower-risk MDS patients — those with very low or low IPSS-R scores — often require only supportive care or low-intensity treatment such as erythropoiesis-stimulating agents for years. Intensive chemotherapy is generally reserved for higher-risk patients or those undergoing transplant preparation. Furthermore, standard intensive chemotherapy regimens used for AML produce responses in MDS but do not improve survival compared with hypomethylating agents in most patient groups. Consequently, treatment intensity should always match disease risk — and overtreatment of low-risk MDS with chemotherapy exposes patients to toxicity without meaningful benefit.
References
- Acute myeloid leukemia (AML) is a bone marrow stem cell cancer that is often fatal despite available treatments.
- ndia has a rich history of women contributing to science. From ancient times to the present, women have made significant strides in various scientific fields.
- MDS World Awareness Day is observed on October 25th each year.
Disclaimer
This article adapts publicly available information from WHO’s Blood Safety and Availability page. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform and not a healthcare provider.
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.