
Aplastic Anemia: When Bone Marrow Stops Making Blood Cells
Every second of every day, the bone marrow performs one of the most vital tasks in the human body. It produces billions of new blood cells — red cells to carry oxygen, white cells to fight infection, and platelets to stop bleeding. This production never stops. It runs continuously throughout life to replace cells that die after their short lifespan.
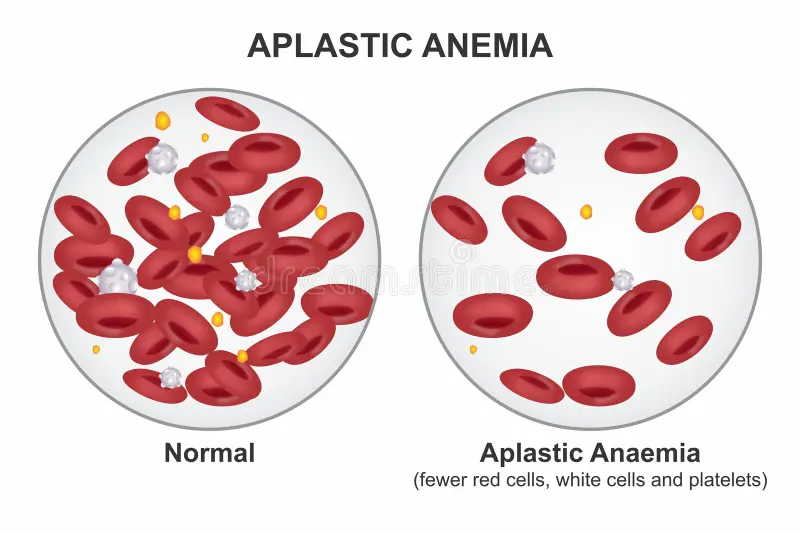
When the bone marrow fails to perform this task, the consequences are profound and potentially fatal. Aplastic anemia is a rare but serious condition in which the bone marrow stops producing enough blood cells of all three types. The result is a body that cannot carry adequate oxygen, cannot fight infection, and cannot stop even minor bleeding.
Aplastic anemia bone marrow stops making blood cells at a rate that quickly depletes the body’s reserves. Without transfusions and definitive treatment, severe aplastic anemia carries a mortality rate exceeding 80% within two years. Furthermore, it can strike at any age — including healthy young adults — making it one of the most devastating diagnoses in haematology. Consequently, understanding what causes bone marrow failure, how it presents, and what modern treatments offer gives patients and families the knowledge they need to navigate this frightening diagnosis with clarity and confidence.
Quick Answer
Aplastic anemia is a rare but life-threatening condition in which bone marrow stops producing enough red blood cells, white blood cells, and platelets. It results from immune destruction or damage to haematopoietic stem cells — the blood-producing cells in the marrow. Treatment includes bone marrow transplantation and immunosuppressive therapy. Early treatment significantly improves survival.
How the Bone Marrow Normally Makes Blood Cells
The Haematopoietic System
Bone marrow fills the hollow spaces inside large bones — including the spine, pelvis, sternum, and long bones of the arms and legs. Within this marrow, haematopoietic stem cells — specialised master cells — continuously divide and differentiate to produce all three types of blood cells. This process is called haematopoiesis — meaning blood cell production.
Red blood cells — called erythrocytes — carry oxygen from the lungs to every tissue in the body through the haemoglobin protein they contain. They live for roughly 120 days before the spleen removes them. White blood cells — called leucocytes — of various types defend the body against bacterial, viral, and fungal infections. Platelets — small cell fragments — circulate in large numbers and rapidly aggregate at sites of blood vessel injury to form clots and stop bleeding. Furthermore, all three cell types have limited lifespans — requiring constant and enormous bone marrow output to maintain adequate circulating numbers.
What Haematopoietic Stem Cells Do
Haematopoietic stem cells are remarkable for two reasons. First, they can self-renew — dividing to produce new stem cells that maintain the stem cell pool indefinitely. Second, they can differentiate — committing to become any type of blood cell the body currently needs. This combination makes them the irreplaceable foundation of the entire blood production system.
When haematopoietic stem cells are destroyed or severely depleted — as happens in aplastic anemia — the marrow loses its ability to regenerate any blood cell type. Consequently, all three blood cell counts fall simultaneously — a pattern called pancytopenia — meaning deficiency of all cell types. It is this simultaneous failure of all blood cell production that makes aplastic anemia so comprehensively dangerous.
What Causes Aplastic Anemia?
Autoimmune Destruction — The Most Common Cause
Aplastic anemia bone marrow stops making blood cells most commonly because the immune system attacks and destroys the haematopoietic stem cells. In roughly 70 to 80% of acquired aplastic anemia cases, autoreactive T lymphocytes — immune cells that normally target foreign threats — mistakenly identify stem cells as foreign and destroy them.
Researchers believe a triggering event — such as a viral infection or drug exposure — activates these autoreactive T cells in genetically susceptible individuals. Once activated, they produce cytokines — inflammatory signalling proteins including interferon-gamma and tumour necrosis factor — that suppress and kill haematopoietic stem cells. The marrow progressively empties as stem cells are destroyed faster than the residual population can self-renew. Consequently, the autoimmune nature of most acquired aplastic anemia explains why immunosuppressive treatment — suppressing the attacking immune cells — produces remission in a significant proportion of patients.
Drug and Chemical Triggers
Certain medications and chemical exposures trigger aplastic anemia in susceptible individuals. The antibiotic chloramphenicol has a well-established association with aplastic anemia. Furthermore, non-steroidal anti-inflammatory drugs, some anticonvulsants — particularly carbamazepine and phenytoin — gold compounds, and several chemotherapy agents all carry documented risks. Benzene — an industrial solvent found in some paints, fuels, and cleaning products — is the most important occupational chemical trigger.
The mechanism varies. Some drugs trigger aplastic anemia through a direct toxic effect on stem cells. Others trigger immune-mediated destruction — activating autoreactive T cells in the same way that viral infections do. Consequently, a thorough medication and occupational exposure history is essential in every newly diagnosed case.
Viral Infections
Several viruses can trigger aplastic anemia. Hepatitis viruses — particularly a non-A non-B non-C hepatitis virus that has not yet been fully characterised — cause a distinctive form called hepatitis-associated aplastic anemia. This pattern accounts for roughly 5% of all aplastic anemia cases and typically affects young men two to three months after an acute hepatitis episode.
Epstein-Barr virus — the cause of infectious mononucleosis — parvovirus B19, cytomegalovirus, and HIV have all been associated with aplastic anemia in some cases. Furthermore, COVID-19 infection has been reported as a trigger in a small number of cases. Consequently, serological testing for these viruses forms part of the standard diagnostic evaluation.
Inherited Causes
A significant minority of aplastic anemia cases — particularly in children and young adults — result from inherited genetic conditions that impair stem cell function or DNA repair. Fanconi anemia is the most common inherited bone marrow failure syndrome. It results from mutations in genes involved in DNA repair — causing stem cells to accumulate DNA damage that eventually triggers cell death or malignant transformation.
Dyskeratosis congenita — caused by mutations affecting telomere maintenance — produces progressive bone marrow failure alongside characteristic skin, nail, and mucosal abnormalities. Diamond-Blackfan anemia, Shwachman-Diamond syndrome, and GATA2 deficiency are additional inherited bone marrow failure syndromes. Consequently, genetic testing is recommended for all younger patients with aplastic anemia — particularly those with dysmorphic features, a positive family history, or unusually short telomeres — because the treatment approach differs significantly from acquired aplastic anemia.
Symptoms of Aplastic Anemia
Symptoms of Low Red Blood Cells
Aplastic anemia bone marrow stops making blood cells in a way that produces three distinct symptom clusters — one for each depleted cell type. Low red blood cell count — called anaemia — produces fatigue and weakness that progressively worsens as haemoglobin falls. The fatigue of aplastic anemia is profound. It is not the tiredness of a poor night’s sleep — it is an exhaustion that limits even minimal physical activity.
Pallor — paleness of the skin, conjunctiva, and mucous membranes — develops as haemoglobin falls below normal levels. Shortness of breath on exertion appears because the blood cannot carry enough oxygen to meet the muscles’ demands during activity. Furthermore, palpitations — an uncomfortable awareness of the heartbeat — develop as the heart compensates for low oxygen delivery by beating faster and harder. Consequently, symptoms of anaemia frequently bring patients to medical attention before other manifestations of aplastic anemia appear.
Symptoms of Low White Blood Cells
Low white blood cell count — called leucopenia — and specifically low neutrophil count — called neutropenia — severely impairs the body’s ability to fight bacterial and fungal infections. Neutrophils are the first-line responders that kill bacteria at sites of infection. Without adequate neutrophil numbers, bacteria and fungi that the healthy immune system controls effortlessly cause overwhelming and life-threatening infections.
Fever — either from opportunistic infection or from the marrow failure itself — is a common presenting symptom. Recurrent mouth ulcers, throat infections, and skin infections occur more frequently than usual. Furthermore, infections that would normally be minor and self-resolving become serious and require hospitalisation. Consequently, any fever in a patient with known or suspected aplastic anemia constitutes a medical emergency requiring urgent assessment and empirical antibiotic treatment without delay. For context on how infections overwhelm compromised immune systems, see our article on septic arthritis — the joint infection that destroys cartilage.
Symptoms of Low Platelets
Low platelet count — called thrombocytopenia — impairs the clotting process and produces bleeding from multiple sites. Easy bruising from minor bumps — appearing as large, purple discolourations on the skin called ecchymoses — is typically the first bleeding manifestation patients notice. Petechiae — tiny, pinpoint red or purple spots caused by microscopic bleeding under the skin — appear over the lower legs and pressure areas.
Prolonged bleeding from minor cuts, bleeding gums, and nosebleeds that are difficult to stop all reflect the reduced platelet count. In severe thrombocytopenia — platelet count below 20,000 per microlitre — the risk of spontaneous internal bleeding rises significantly. Intracranial haemorrhage — bleeding inside the skull — is the most feared complication and can be fatal. Consequently, maintaining platelet counts above safe thresholds through transfusion is a critical component of supportive care while definitive treatment is established.
How Doctors Diagnose Aplastic Anemia
Blood Tests and Initial Findings
Diagnosing aplastic anemia bone marrow stops making blood cells begins with a full blood count. The characteristic finding is pancytopenia — simultaneous reduction in red blood cells, white blood cells, and platelets. Haemoglobin falls below normal — sometimes severely. The absolute neutrophil count — a critical measure of infection-fighting capacity — falls below normal and in severe cases drops below 500 cells per microlitre. Platelet count falls well below the normal range of 150,000 to 400,000 per microlitre.
The blood film — microscopic examination of a thin smear of blood — provides important additional information. In aplastic anemia, the remaining cells look morphologically normal — they are simply reduced in number. This normality distinguishes aplastic anemia from conditions where abnormal-looking cells replace normal ones — such as leukaemia — or where abnormal red cell shapes suggest a haemolytic process. Consequently, the blood film is an essential part of distinguishing aplastic anemia from its mimics.
Bone Marrow Biopsy
Bone marrow biopsy — extracting a sample of bone marrow tissue from the posterior iliac crest of the pelvis under local anaesthetic — is the definitive diagnostic investigation. It directly demonstrates the characteristic finding of aplastic anemia — a hypocellular marrow in which the normal haematopoietic cells have been replaced by fat cells.
In a healthy adult, bone marrow is roughly 30 to 70% cellular — packed with active haematopoietic cells producing blood cells. In aplastic anemia, cellularity falls below 25% — and in severe cases below 10% — leaving a marrow that is mostly empty fat with only scattered residual blood-forming cells. Furthermore, the biopsy excludes other causes of pancytopenia — particularly marrow infiltration by leukaemia, lymphoma, myeloma, or metastatic cancer — that would require completely different treatment. Consequently, bone marrow biopsy is mandatory before any treatment begins.
Severity Classification
Aplastic anemia receives severity classification that directly guides treatment intensity. Severe aplastic anemia — called SAA — requires meeting two of three blood count criteria: neutrophil count below 500 cells per microlitre, platelet count below 20,000 per microlitre, or reticulocyte count below 20,000 per microlitre — combined with a marrow cellularity below 25%. Very severe aplastic anemia — called vSAA — is defined by a neutrophil count below 200 cells per microlitre.
Non-severe aplastic anemia — sometimes called moderate aplastic anemia — does not meet the criteria for severe disease. It produces pancytopenia but with higher residual blood counts. Consequently, treatment urgency is highest for very severe aplastic anemia — where the risk of fatal infection or bleeding is immediate — and less urgent for non-severe disease where watchful waiting with close monitoring is sometimes appropriate.
Treatment of Aplastic Anemia
Supportive Care
Treatment of aplastic anemia bone marrow stops making blood cells begins with supportive care — managing the immediate life-threatening consequences of pancytopenia while definitive treatment is arranged. Red blood cell transfusions correct symptomatic anaemia and maintain haemoglobin at levels that allow adequate daily function. Platelet transfusions prevent or treat dangerous bleeding — particularly when platelet counts fall below 10,000 per microlitre.
Broad-spectrum antibiotics and antifungal medications prevent and treat the opportunistic infections that the neutropenic patient cannot fight independently. Growth factors — particularly granulocyte colony-stimulating factor, called G-CSF — stimulate residual neutrophil production and help maintain infection-fighting capacity during the period before definitive treatment takes effect. Furthermore, strict infection prevention measures — including protective isolation, meticulous hand hygiene, and avoidance of raw foods — reduce infection risk in profoundly neutropenic patients. Consequently, supportive care bridges the gap between diagnosis and definitive treatment.
Bone Marrow Transplantation
Bone marrow transplantation — more precisely called allogeneic haematopoietic stem cell transplantation — is the only curative treatment for aplastic anemia. It replaces the patient’s destroyed or dysfunctional stem cells with healthy donor stem cells capable of regenerating the entire blood production system permanently.
The best outcomes come from a matched sibling donor — a brother or sister who shares the same HLA tissue type as the patient. Matched sibling donor transplantation in young patients with severe aplastic anemia achieves long-term cure rates exceeding 90% in experienced transplant centres. Furthermore, transplantation in young patients eliminates the risk of relapse and transformation to myelodysplastic syndrome or leukaemia — risks that persist indefinitely after immunosuppressive therapy.
When a matched sibling donor is unavailable, a matched unrelated donor — found through international bone marrow registries — provides an alternative. Matched unrelated donor transplantation carries higher risks of graft-versus-host disease — a serious complication in which donor immune cells attack the recipient’s tissues — and produces slightly lower cure rates than sibling transplantation. Consequently, matched sibling donor transplantation is the first-line treatment of choice for younger patients with severe aplastic anemia. For context on how immunological mismatches cause tissue damage, see our article on lupus nephritis — when lupus attacks the kidneys.
Immunosuppressive Therapy
For patients who lack a suitable transplant donor, who are too old or medically unfit for transplantation, or who have non-severe aplastic anemia not requiring immediate transplantation, immunosuppressive therapy targets the autoreactive T cells destroying haematopoietic stem cells.
The standard immunosuppressive regimen combines antithymocyte globulin — called ATG — with cyclosporine. Antithymocyte globulin is a preparation of antibodies derived from horses or rabbits that depletes the T lymphocytes attacking the marrow. Horse ATG combined with cyclosporine achieves haematological response — sufficient recovery of blood cell production to allow transfusion independence — in roughly 60 to 70% of patients. Furthermore, the addition of eltrombopag — a thrombopoietin receptor agonist that stimulates stem cell proliferation — to the standard ATG and cyclosporine regimen has improved response rates to over 80% in recent clinical trials.
Cyclosporine continues for at least twelve to twenty-four months after ATG to maintain remission and prevent relapse. Furthermore, regular blood count monitoring during this period detects early relapse — allowing prompt retreatment before the marrow fails again completely. Consequently, patients who achieve remission with immunosuppressive therapy require sustained medical follow-up for several years after treatment.
Managing Relapse and Clonal Evolution
Relapse after immunosuppressive therapy — recurrence of aplastic anemia after an initial response — occurs in roughly 30% of patients. Many patients respond to repeat immunosuppression. Furthermore, eltrombopag monotherapy or second-line immunosuppressive agents — including cyclophosphamide and alemtuzumab — are options for patients who fail multiple treatment courses.
A more serious long-term complication of aplastic anemia — particularly after immunosuppressive treatment — is clonal evolution. This refers to the development of a related but distinct blood disorder — including myelodysplastic syndrome, paroxysmal nocturnal haemoglobinuria, or acute myeloid leukaemia — in a proportion of patients over years. Consequently, long-term haematological surveillance with annual blood counts, blood film review, and periodic bone marrow assessment is essential for all aplastic anemia survivors.
Living With Aplastic Anemia
During Active Treatment
Active treatment for aplastic anemia — whether transplantation or immunosuppressive therapy — places significant physical and psychological demands on patients and their families. Transplant recipients spend weeks in hospital during engraftment — the period when donor stem cells travel to the marrow and begin producing blood cells. Furthermore, the profound immunosuppression required for transplantation preparation increases infection risk to its highest level.
Patients undergoing immunosuppressive therapy experience a period of weeks to months before blood counts recover adequately. During this period, regular clinic attendance for blood count monitoring, transfusion support, and infection management is necessary. Moreover, activity restrictions — avoiding crowds, contact sports, and other high-risk situations — protect neutropenic and thrombocytopenic patients from avoidable harm. Consequently, family support, psychological counselling, and clear patient education significantly improve coping and treatment adherence during this demanding phase.
Long-Term Health After Remission
Patients who achieve remission after transplantation or immunosuppressive therapy can return to normal life — including work, study, exercise, and family planning — in the majority of cases. However, long-term monitoring remains essential to detect relapse, clonal evolution, and the late complications of treatment. These include secondary malignancies — particularly in patients who received radiation-containing conditioning regimens before transplantation — and bone density loss from prolonged corticosteroid use. For context on managing bone health in patients with chronic conditions requiring long-term treatment, see our article on osteoporosis — how bones lose density and what reverses it.
Furthermore, patients who received transplantation remain on immunosuppressive medications for months to years post-transplant to prevent graft-versus-host disease. These medications increase infection risk and require regular monitoring of drug levels, kidney function, and blood pressure. Consequently, a long-term relationship with a specialist haematology team is essential for all aplastic anemia survivors.
When to Seek Urgent Medical Help
Seek emergency medical care immediately if a person with known or suspected aplastic anemia develops fever above 38 degrees Celsius — which may indicate a life-threatening infection in a neutropenic patient. Furthermore, seek emergency care for any spontaneous bleeding — particularly nosebleeds that will not stop after thirty minutes, unexplained large bruises, blood in the urine or stool, or sudden severe headache — which may indicate intracranial bleeding.
Consequently, patients with aplastic anemia and their families must have a clear, practised emergency plan for recognising and responding to these warning signs without delay. Every hour matters when infection or bleeding strikes a patient without adequate white cells or platelets.
Frequently Asked Questions
1. Is aplastic anemia the same as leukaemia?
No. Aplastic anemia and leukaemia are distinct conditions with very different mechanisms. Aplastic anemia involves bone marrow failure — the marrow stops producing blood cells and becomes empty. Leukaemia involves uncontrolled proliferation of abnormal blood cells — the marrow becomes packed with cancerous cells that crowd out normal production. Furthermore, treatment approaches are different — aplastic anemia requires restoration of normal stem cells through transplantation or immunosuppression, while leukaemia requires elimination of the malignant clone through chemotherapy. Consequently, accurate diagnosis through bone marrow biopsy is essential before treatment begins.
2. Can aplastic anemia be cured?
Yes. Bone marrow transplantation from a matched sibling donor achieves long-term cure in over 90% of young patients with severe aplastic anemia. Furthermore, immunosuppressive therapy produces durable remission in a significant proportion of patients who are not transplant candidates. However, immunosuppressive therapy carries a risk of relapse and clonal evolution that transplantation eliminates. Consequently, transplantation is the preferred curative treatment whenever a suitable donor and a medically fit patient align.
3. How is aplastic anemia diagnosed?
Aplastic anemia is diagnosed through a full blood count showing pancytopenia — simultaneous reduction in all three blood cell types — confirmed by bone marrow biopsy showing a hypocellular marrow with replacement of normal haematopoietic cells by fat cells. Furthermore, additional investigations exclude other causes of pancytopenia — including leukaemia, vitamin deficiencies, and inherited bone marrow failure syndromes. Consequently, bone marrow biopsy is mandatory for all patients with suspected aplastic anemia before any treatment begins.
4. Is aplastic anemia hereditary?
Most cases of aplastic anemia are acquired — resulting from immune-mediated destruction triggered by infections, drug exposures, or unknown factors — rather than inherited. However, inherited bone marrow failure syndromes including Fanconi anemia, dyskeratosis congenita, and others cause aplastic anemia in a minority of patients — particularly children and young adults. Furthermore, identifying an inherited cause changes the treatment approach — for example, Fanconi anemia requires modified transplant conditioning to avoid excessive toxicity. Consequently, genetic testing is recommended for all younger patients with aplastic anemia.
5. What is the difference between aplastic anemia and other types of anemia?
Most other anemias involve a deficiency of red blood cells alone — from iron deficiency, vitamin B12 deficiency, haemolysis, or chronic disease. Aplastic anemia differs fundamentally because it involves simultaneous deficiency of all three blood cell types — red cells, white cells, and platelets — reflecting a failure of the entire bone marrow production system rather than a single cell lineage. Furthermore, treating aplastic anemia with iron or vitamin supplements — appropriate for other anemias — produces no benefit whatsoever. Consequently, recognising the pancytopenic pattern and pursuing bone marrow investigation distinguishes aplastic anemia from these other more common conditions.
References
- When the 77th World Health Assembly convened in Geneva in May 2024, Spain spearheaded a resolution that would reshape global transplantation policy for the next decade.
- The Council of Scientific & Industrial Research – National Institute of Science Communication and Policy Research
- The Anemia Mukt Bharat strategy employs a life cycle approach to tackle anemia, focusing on various demographics.
Disclaimer
This article adapts publicly available information from WHO’s Blood Safety and Availability page. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform and not a healthcare provider.
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.