
Myasthenia Gravis: The Autoimmune Disease That Makes Muscles Tire Rapidly
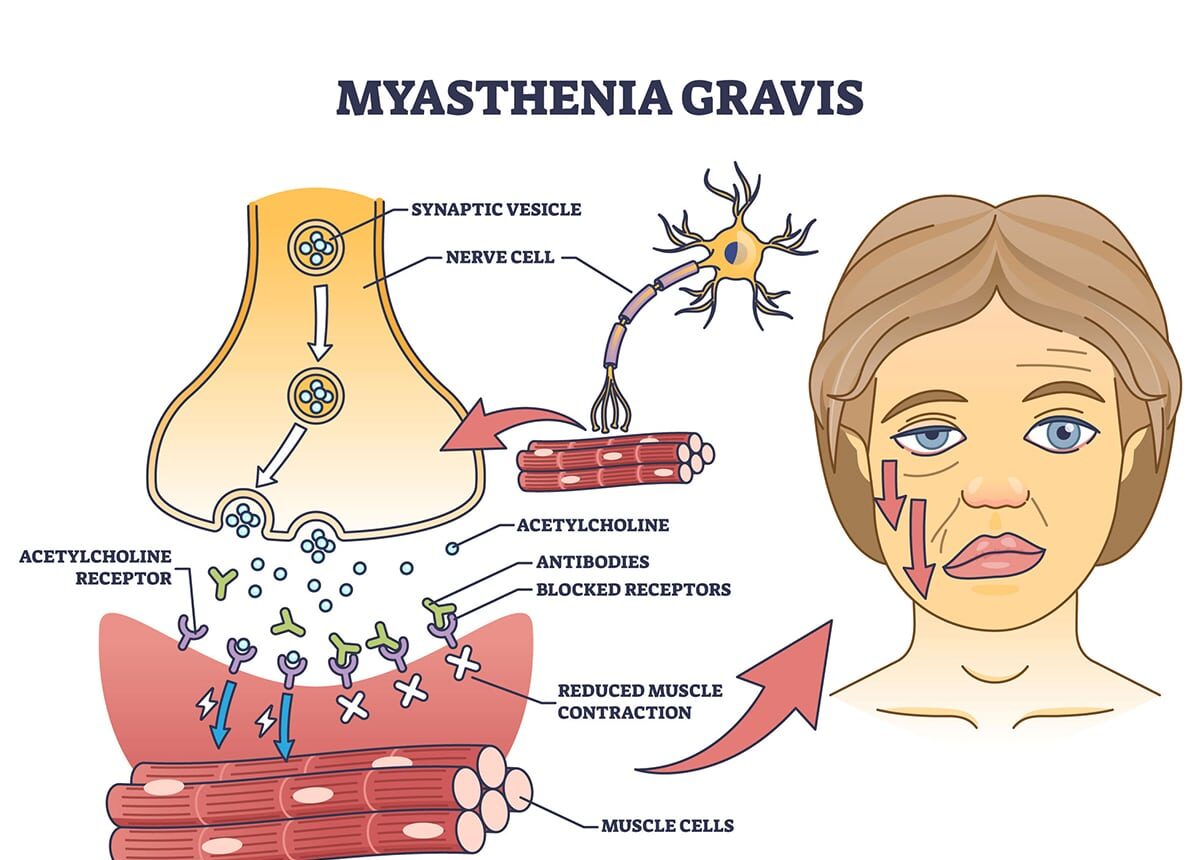
When 28-year-old Priya developed drooping eyelids and double vision that worsened throughout the day but improved after rest, her ophthalmologist suspected myasthenia gravis (MG)—a chronic autoimmune neuromuscular disorder affecting approximately 15-20 per 100,000 people worldwide, caused by antibodies attacking the communication junction between nerves and muscles, specifically targeting acetylcholine receptors or related proteins at the neuromuscular junction where nerve signals tell muscles to contract. Her neurologist explained that in myasthenia gravis, the immune system mistakenly produces antibodies that block, alter, or destroy these crucial receptors, dramatically reducing the number of available receptors and weakening the nerve-to-muscle signal, resulting in the hallmark feature of fluctuating muscle weakness and abnormal muscle fatigue where muscles work fine initially but tire rapidly with repeated use, typically affecting eye muscles first (causing drooping eyelids and double vision in 85% of patients as initial symptoms), then spreading to facial muscles, throat muscles (causing difficulty swallowing, chewing, and speaking), and limb muscles, with symptoms characteristically worsening with activity and improving with rest. Understanding myasthenia gravis is crucial because it’s often misdiagnosed initially as conditions ranging from stroke to simple tiredness, yet specific diagnostic tests (antibody tests, electrophysiology testing, and the dramatic temporary improvement with edrophonium injection—the “Tensilon test”) can confirm diagnosis, the disease can be life-threatening when respiratory muscles are affected (myasthenic crisis requiring emergency ventilation support), yet with modern treatment including cholinesterase inhibitors that improve neuromuscular transmission, immunosuppressive medications that reduce antibody production, thymectomy (surgical removal of the thymus gland) which leads to improvement in many patients, and newer biological therapies targeting specific immune pathways, most patients achieve good symptom control and near-normal life expectancy, though lifelong management is typically required.
The Neuromuscular Junction and Acetylcholine Receptors: When Communication Breaks Down
Myasthenia gravis is fundamentally a disease of the neuromuscular junction—the specialized synapse where motor neurons (nerve cells) communicate with skeletal muscle fibers to initiate contraction. Understanding normal neuromuscular transmission helps explain what goes wrong in MG. In normal neuromuscular function, when a motor neuron sends a signal down its axon to the muscle, the nerve terminal releases acetylcholine (a neurotransmitter) into the synaptic cleft (narrow gap between nerve and muscle). Acetylcholine molecules diffuse across the cleft and bind to acetylcholine receptors (AChRs) densely clustered on the muscle membrane at the motor endplate. Binding triggers opening of ion channels, allowing sodium to rush into the muscle fiber, creating an electrical potential that spreads across the muscle membrane and triggers muscle contraction. The enzyme acetylcholinesterase then breaks down acetylcholine, terminating the signal and preparing for the next transmission. This process occurs within milliseconds, allowing rapid, precise muscle control.
In myasthenia gravis, autoantibodies (antibodies directed against one’s own tissues) attack components of the neuromuscular junction, disrupting this communication. The most common pattern (80-85% of MG patients) involves anti-AChR antibodies binding to acetylcholine receptors on the muscle membrane, which blocks acetylcholine from binding (competitive inhibition), causes receptors to be internalized and destroyed (accelerated degradation), activates complement (immune proteins that damage the motor endplate), and ultimately reduces the number of functional AChRs by 70-90% in severely affected muscles. The muscle membrane is literally less sensitive to acetylcholine due to fewer receptors.
Another pattern (5-8% of MG patients) shows anti-MuSK antibodies (muscle-specific kinase) targeting MuSK protein, which is essential for clustering AChRs at the neuromuscular junction. Anti-MuSK MG tends to affect face, bulbar (throat/swallowing), and respiratory muscles more than limb muscles and often has more severe symptoms and responds differently to treatments. A third pattern (1-5% of patients) involves antibodies against LRP4 (lipoprotein receptor-related protein 4), another protein involved in AChR clustering. About 5-10% of MG patients are “seronegative” (antibody-negative)—they have clinical MG but standard antibody tests are negative. Some have low-level antibodies below detection, others may have antibodies against currently unknown targets.
The result of reduced functional receptors is called the “safety factor” reduction. Normally, neuromuscular transmission has a large safety margin—more acetylcholine is released and more receptors are available than strictly necessary for contraction. In MG, this safety factor is reduced or lost. The first few muscle contractions work reasonably well because enough acetylcholine accumulates despite fewer receptors, but with repeated use, acetylcholine release diminishes slightly with each signal (normal physiology), and with fewer receptors available, the reduced acetylcholine can no longer activate enough receptors to generate muscle contraction. This causes the characteristic fatigable weakness—muscles work initially but tire rapidly.
What causes the autoimmune attack is not fully understood, but the thymus gland (located behind the breastbone) plays a critical role. The thymus normally trains T-cells (immune cells) to distinguish self from non-self during development. About 75% of MG patients have thymic abnormalities including thymic hyperplasia (overactive thymus with germinal centers—areas of antibody production) in 60-70%, particularly in younger patients, and thymoma (thymus tumor) in 10-15%, more common in older patients. The thymus likely generates or perpetuates the anti-AChR immune response, explaining why thymectomy (surgical removal) improves many patients. MG can occur at any age but shows bimodal distribution—peaks in young women (ages 20-40) and older men (ages 60-80). Female-to-male ratio is about 3:2 in early-onset MG, but roughly equal or male-predominant in late-onset MG.
Symptoms: Fluctuating Weakness With Characteristic Patterns
Myasthenia gravis symptoms are highly distinctive once recognized, though initially subtle and easily missed. The hallmark features include muscle weakness that worsens with repeated use (fatigability), improves with rest, and fluctuates throughout the day (often worse in evening after a day’s activities). Ocular symptoms occur as the initial presentation in 85% of patients, with ptosis (drooping eyelids—often asymmetric, one eye more than the other) worsening with sustained upward gaze and improving after closing eyes briefly. Diplopia (double vision) occurs, particularly with lateral gaze (looking to sides) or reading, and often varies—sometimes horizontal, sometimes vertical. About 15% of patients remain with pure ocular MG never progressing beyond eye symptoms.
Bulbar symptoms affect face, throat, and jaw muscles with facial weakness causing weak smile (transverse smile or “snarl”), difficulty puffing cheeks or whistling, and expressionless face. Dysarthria (speech difficulties) shows nasal voice quality, slurred speech, voice fading during long conversations, and difficulty with sustained talking. Dysphagia (swallowing difficulty) brings trouble swallowing solids first, then liquids, nasal regurgitation (liquids coming through nose when swallowing), choking on food or liquids, and risk of aspiration pneumonia. Chewing fatigue causes jaw muscles tiring while eating, needing to rest mid-meal, and preferring soft foods requiring less chewing.
Limb weakness typically develops later and is proximal predominant (shoulders, hips more than hands/feet), with difficulty lifting arms overhead (brushing hair, reaching high shelves), difficulty standing from sitting or climbing stairs, upper extremities more affected than lower, and weakness usually symmetric (both sides equally). Respiratory muscle weakness is the most serious manifestation with shortness of breath, particularly when lying flat, weak cough, difficulty taking deep breaths, and orthopnea (needing to sleep propped up). Myasthenic crisis represents severe generalized weakness with respiratory failure requiring emergency intubation and mechanical ventilation—occurs in 15-20% of patients at some point and is triggered by infections, surgery, certain medications, stress, or sometimes no identifiable trigger.
The fluctuating pattern is pathognomonic (highly characteristic) of MG, with symptoms worse later in the day after sustained muscle use, improved in the morning after sleep, worsening with heat (hot weather, hot baths), and improved with cold. Symptoms can vary day-to-day unpredictably. Certain features help distinguish MG from other conditions: purely motor symptoms with no sensory changes (no numbness, tingling, pain), normal reflexes initially (may be reduced in severe weakness), and no muscle atrophy until very late (unlike motor neuron diseases). Preserved pupillary responses to light distinguish MG from other causes of ptosis and diplopia.
MG is classified by severity using the Myasthenia Gravis Foundation of America (MGFA) classification: Class I is ocular symptoms only, Class II shows mild generalized weakness (IIa predominantly limb/trunk, IIb predominantly bulbar/respiratory), Class III has moderate generalized weakness, Class IV involves severe generalized weakness, Class V is defined by intubation with or without mechanical ventilation (myasthenic crisis), and pharmacologic remission means asymptomatic on medications. Many patients progress from ocular to generalized MG within 2 years, though some remain ocular indefinitely.
Diagnosis: Clinical Recognition and Confirmatory Testing
Diagnosing myasthenia gravis requires clinical suspicion based on the characteristic pattern of fluctuating fatigable weakness, confirmed by specific tests. Clinical examination includes the “curtain sign”—ptosis worsening after sustained upward gaze for 30-60 seconds, the “sleep test” or “ice pack test”—ptosis improving after 2-5 minutes of closed eyes (rest) or ice pack on closed eyes (cold improves NMJ transmission), fatigue on repetitive testing such as counting aloud to 50 (voice becomes nasal/weak), repetitive arm raising (weakness develops), and asking patient to look laterally continuously (diplopia develops).
Bedside pharmacologic testing uses the Tensilon test (edrophonium test)—historically diagnostic, less commonly used now. Edrophonium is a short-acting cholinesterase inhibitor given IV, blocking acetylcholine breakdown and temporarily increasing acetylcholine at the NMJ. In MG, dramatic improvement occurs within 30-60 seconds (drooping lids lift, double vision resolves, strength improves). Effect lasts only 5-10 minutes. The test requires cardiac monitoring (edrophonium can cause bradycardia) and atropine available as antidote. False positives and negatives occur, so other tests are preferred now.
Serologic (antibody) testing provides confirmation with anti-AChR antibodies positive in 80-85% of generalized MG, 50% of ocular MG (lower sensitivity in pure ocular disease). Highly specific—if positive, confirms MG diagnosis. Anti-MuSK antibodies are found in 5-8% of MG patients, typically seronegative for AChR. Suggests more severe bulbar and respiratory involvement. Anti-LRP4 antibodies occur in 1-5%, usually in double-seronegative patients (negative for both AChR and MuSK). About 5-10% remain seronegative despite clinical MG—diagnosis based on clinical features and electrophysiology.
Electrodiagnostic testing includes repetitive nerve stimulation (RNS)—motor nerves stimulated at 2-3 Hz repeatedly while recording muscle electrical response. In MG, >10% decrement (reduction) in response amplitude between first and fourth/fifth stimulus indicates NMJ transmission failure. Sensitivity is only 50-75% (many false negatives), but high specificity when positive. Single-fiber electromyography (SFEMG) measures “jitter” (variability in timing of muscle fiber activation). Increased jitter indicates NMJ transmission instability. Most sensitive test for MG (95-99% sensitivity), though less specific (abnormal in other NMJ disorders too).
Imaging and additional testing involve chest CT or MRI looking for thymoma (found in 10-15% of MG patients)—all MG patients should have thymic imaging. Checking thyroid function since thyroid disease commonly coexists with MG (both autoimmune), and pulmonary function tests measuring vital capacity and inspiratory/expiratory pressures assessing respiratory muscle strength (critical for monitoring and predicting crisis risk).
Differential diagnosis includes other conditions causing ptosis/ophthalmoplegia such as cranial nerve palsies (third, fourth, sixth nerve), brainstem stroke (but would have sensory findings and abnormal pupils), chronic progressive external ophthalmoplegia (mitochondrial—but slowly progressive without fluctuation), and Lambert-Eaton myasthenic syndrome (LEMS—presynaptic NMJ disorder from antibodies against voltage-gated calcium channels; weakness improves with exercise initially versus worsening in MG). Other neuromuscular conditions to consider are motor neuron disease like ALS (progressive without fluctuation, has fasciculations and atrophy), muscular dystrophies (progressive chronic weakness without fatigability), and inflammatory myopathies like polymyositis (elevated CK, muscle pain, different pattern).
Treatment: Symptomatic and Immunomodulatory Approaches
Myasthenia gravis treatment has multiple components addressing both symptoms and underlying autoimmune process. Symptomatic treatment uses cholinesterase inhibitors with pyridostigmine (Mestinon) as the mainstay first-line treatment improving muscle strength by increasing acetylcholine availability at the NMJ. It doesn’t affect underlying disease but improves symptoms. Typical dosing is 60 mg every 4-6 hours, adjusted based on response. Benefits include rapid improvement (30-60 minutes), no immunosuppression (safer than immunosuppressive drugs), and effectiveness for mild-moderate symptoms. Limitations show incomplete symptom control in most patients (rarely adequate as monotherapy), and side effects from excess acetylcholine including GI cramps, diarrhea, salivation, and muscle twitching. Severe overdose can cause cholinergic crisis (weakness from too much acetylcholine—mimics myasthenic crisis). It provides symptomatic relief but isn’t disease-modifying, so most patients need additional immunomodulatory treatment.
Immunosuppressive therapy reduces antibody production using corticosteroids. Prednisone is often first-line immunosuppression with initial worsening possible in the first 7-10 days (transient weakness before improvement—hospitalization sometimes needed during initiation), then improvement beginning at 2-4 weeks and maximum benefit at 6-12 months. Long-term steroids cause significant side effects (weight gain, osteoporosis, diabetes, cataracts, mood changes). Steroid-sparing agents are used to reduce steroid doses, with azathioprine (Imuran) taking 6-12 months for full effect and being effective for long-term maintenance with good safety profile. Mycophenolate mofetil (CellCept) works similarly to azathioprine as an alternative, while methotrexate is used less commonly, and cyclosporine and tacrolimus (calcineurin inhibitors) are reserved for refractory cases given toxicity.
Thymectomy (surgical thymus removal) is recommended for patients with thymoma (mandatory—thymomas can be malignant) and generalized MG under age 65 with AChR antibodies (even without thymoma). Benefits include improvement in 70-80% of patients over 2-5 years post-surgery, with 15-20% achieving complete remission off all medications and 50-60% significantly improving allowing medication reduction. The mechanism isn’t fully understood (removing source of antibody production and abnormal immune stimulation), and improvements take years (not immediate—continue medications post-surgery). Surgery can be performed via median sternotomy (traditional—splitting breastbone), thoracoscopy (minimally invasive—multiple small incisions), or robotic-assisted thoracoscopy.
Rapid immunomodulation for severe exacerbations or myasthenic crisis includes plasmapheresis (plasma exchange)—physically removing antibodies from blood, effective within days, and used for acute severe weakness or pre-operatively. Effects last 3-6 weeks (temporary—antibodies return). Intravenous immunoglobulin (IVIg) involves infusing pooled antibodies from donors, neutralizing pathogenic antibodies through unknown mechanisms, and showing effectiveness within 1-2 weeks lasting 3-6 weeks (temporary). Both plasmapheresis and IVIg provide rapid temporary improvement—bridge therapies while waiting for immunosuppression to work or for crisis management.
Newer biologic therapies include eculizumab (Soliris)—monoclonal antibody blocking complement protein C5, FDA-approved 2017 for refractory generalized MG, showing significant benefit in clinical trials but requiring every-2-week IV infusions. It’s extremely expensive ($400,000+ annually) and increases meningococcal infection risk (requiring vaccination). Rituximab (anti-CD20 antibody) depletes B-cells (antibody-producing cells) and shows effectiveness particularly in anti-MuSK MG, though it’s used off-label. Emerging therapies include various immunomodulators targeting different immune pathways in clinical trials.
Management of myasthenic crisis is a medical emergency requiring ICU admission with intubation and mechanical ventilation when respiratory failure develops. Plasmapheresis or IVIg is given immediately (faster effect than steroids), with high-dose IV steroids, stopping cholinesterase inhibitors temporarily (differentiate myasthenic from cholinergic crisis), treating precipitating factors (infections, etc.), and close monitoring for weaning from ventilator.
Medication precautions are critical since certain drugs can worsen MG and trigger crisis, including aminoglycoside antibiotics (gentamicin, tobramycin), fluoroquinolones (ciprofloxacin, levofloxacin), macrolides (azithromycin, erythromycin), beta-blockers, calcium channel blockers, magnesium (including IV magnesium for pre-eclampsia), certain anesthetics (succinylcholine, curare-type agents), and quinine/quinidine. Patients should always inform healthcare providers about MG diagnosis before any procedure or new medication.
Living with Myasthenia Gravis: Long-Term Outlook and Quality of Life
Living with myasthenia gravis means managing a chronic autoimmune condition requiring lifelong treatment in most cases, though the prognosis has improved dramatically with modern therapies. Before the 1960s (prior to modern immunosuppression and ventilatory support), mortality was 30-40%, with most deaths from respiratory failure. With current treatment, mortality is less than 5%, and life expectancy approaches normal for most patients. However, MG significantly impacts quality of life through unpredictable symptom fluctuations creating uncertainty about daily function—patients may feel relatively well one day and very weak the next. Medication side effects from chronic immunosuppression cause weight gain, bone loss, increased infection risk, and other complications. Physical limitations vary from mild restrictions to significant disability depending on severity. Employment challenges arise when jobs requiring sustained visual attention, speaking, or physical activity become difficult.
Disease course varies enormously between patients. Some achieve complete stable remission (15-20%) being symptom-free off all medications for years—most after thymectomy plus immunosuppression. Pharmacologic remission occurs in 30-40% who remain asymptomatic but require ongoing low-dose medications. Improved but symptomatic patients (30-40%) have good symptom control with medications but ongoing mild-moderate symptoms and medication side effects. Refractory MG affects 10-15% who have poor response to standard treatments, requiring multiple immunosuppressants and/or biologics with significant disability.
Pregnancy considerations show MG can worsen, improve, or stay stable during pregnancy (unpredictable). About one-third experience each pattern. Pyridostigmine is safe in pregnancy. Prednisone and azathioprine are generally considered acceptable. Avoid methotrexate and mycophenolate (teratogenic). Plasmapheresis and IVIg are safe if needed. Delivery planning is important since pushing during labor can be exhausting for weak patients (assisted delivery sometimes needed). Magnesium sulfate (used for pre-eclampsia/eclampsia) can precipitate crisis—must be avoided. Neonatal myasthenia occurs in 10-20% of babies born to MG mothers as maternal antibodies cross placenta, causing temporary weakness in newborn (feeding difficulties, weak cry, hypotonia). This resolves as maternal antibodies clear over 2-4 weeks and requires supportive care (feeding support, rarely temporary ventilation).
Psychosocial impact includes anxiety and depression common given chronic disease, unpredictable symptoms, and medication burdens. Fatigue beyond muscle weakness causes disabling exhaustion even when muscles seem relatively strong. Social isolation happens from unpredictable symptoms limiting ability to commit to plans, visible symptoms (ptosis, slurred speech) causing self-consciousness, and inability to participate in previous activities. Relationship challenges emerge with family/partners needing to adapt to disease and provide support, some relationships not surviving the stress, and concerns about being a burden.
Support resources include the Myasthenia Gravis Foundation of America providing education, support groups, research funding, and patient advocacy. Muscular Dystrophy Association clinics offer comprehensive care for MG patients. Online communities and local support groups connect patients for emotional support and practical advice. Disability resources and workplace accommodations help with employment challenges.
Tips for living well with MG involve pacing activities and planning rest periods throughout the day, avoiding triggers like heat, infections, stress, and certain medications, maintaining healthy lifestyle with good nutrition and gentle regular exercise, staying current with vaccinations (flu, pneumonia, COVID-19), communicating openly with family, friends, employers about limitations, wearing medical alert bracelet noting MG and crisis risk, keeping emergency plan written clearly with medications, crisis symptoms, and emergency contacts, and staying connected to MG community and care team.
Frequently Asked Questions
Q1: I was just diagnosed with myasthenia gravis. My main symptom is drooping eyelids. Will this definitely spread to other muscles, and is there anything I can do to prevent it?
Your question reflects a common concern for patients with newly diagnosed ocular myasthenia gravis. The progression pattern varies significantly between patients. About 50-80% of patients who present with pure ocular symptoms will progress to generalized MG (involving other muscle groups beyond the eyes) within 2 years of symptom onset, with most progression occurring in the first year. About 85% who will generalize do so within 3 years. However, this means 15-20% remain with pure ocular MG indefinitely, never developing generalized disease. Unfortunately, we cannot reliably predict which patients will progress and which will remain ocular, though some factors suggest higher risk for generalization including younger age at onset (under 40), anti-AChR antibodies positive at high titers, and abnormal repetitive nerve stimulation or SFEMG testing beyond ocular muscles. Factors suggesting better chance of remaining ocular include older age at onset (over 50), isolated ocular symptoms for more than 2 years already, and negative antibody testing (seronegative ocular MG less likely to generalize).
Regarding prevention of generalization, the evidence is debated. Early immunosuppressive treatment (corticosteroids) may reduce the risk of progression from ocular to generalized MG—some studies suggest starting prednisone early in ocular MG reduces generalization risk, though other studies are less convincing. The decision involves weighing potential benefit (possibly preventing generalization) against definite risks (steroid side effects over potentially decades if you remain ocular). Many neurologists don’t routinely treat pure ocular MG with steroids unless symptoms are severe and affecting quality of life (significant ptosis interfering with vision, bothersome diplopia). Instead, they use pyridostigmine for symptom control and monitor closely for signs of generalization. If you have only mild ptosis not bothering you much, observation may be reasonable. If symptoms are bothersome, treatment options include pyridostigmine first—often helps ocular symptoms though incompletely, with doses up to 60 mg every 4 hours if needed. If inadequate, prednisone can be considered, typically started at low-moderate doses (20-40 mg daily), with possible temporary worsening in the first week, then gradual improvement over weeks to months. Some physicians use alternate-day dosing to minimize side effects.
Thymectomy is generally not recommended for pure ocular MG without generalization, though if thymic imaging shows thymoma, surgery is required regardless. Other considerations include having baseline pulmonary function tests to establish respiratory muscle strength before any progression and learning warning signs of generalization including difficulty swallowing, chewing fatigue, voice changes, limb weakness, or breathing problems. Regular follow-up every 3-6 months initially is important, especially in the first 2 years when generalization risk is highest. If you remain purely ocular for 2-3 years, the likelihood of staying ocular increases significantly. The emotional aspect matters—uncertainty about progression is stressful. Some patients find it helpful to prepare mentally for possible generalization while hoping to remain ocular, knowing that if generalization occurs, it’s treatable. The outlook for treated generalized MG is good for most patients.
What I’d recommend is discussing risks versus benefits of early immunosuppression with your neurologist, considering your age, antibody status, severity of current symptoms, and personal preferences. If symptoms are mild and you wish to avoid steroids, close monitoring with pyridostigmine for symptom control is reasonable. If symptoms are bothersome or you’re very anxious about progression, early prednisone might be considered. Regardless, close follow-up and awareness of warning signs are essential in the first 1-2 years.
Q2: I’m on pyridostigmine and prednisone for myasthenia gravis, but I still have some weakness and fatigue. My doctor says I’m “doing well,” but I don’t feel well. Should I expect to feel completely normal, or is some persistent weakness just part of having MG?
Your frustration is completely understandable and very common among MG patients. There’s often a disconnect between physician assessment of “doing well” (meaning not in crisis, significant objective improvement from baseline, function maintained) and patient experience of ongoing symptoms affecting quality of life. The reality of MG treatment expectations requires some honest discussion. Complete symptom elimination is uncommon—only about 15-20% of MG patients achieve complete stable remission (no symptoms off all medications). Most patients (60-70%) achieve “pharmacologic remission” or “minimal manifestations” meaning minimal or mild symptoms on medications allowing relatively normal function but not feeling completely “normal.” About 10-15% remain significantly symptomatic despite multiple treatments (refractory MG).
Persistent mild symptoms are typical even with good treatment response. Many treated MG patients experience residual mild ptosis or diplopia (particularly late in the day or with prolonged visual tasks), some facial weakness or dysarthria with prolonged talking, mild dysphagia with certain foods (thick or dry foods harder to swallow), some limb weakness with sustained activities, and fatigue disproportionate to objective weakness (this is very common and not fully explained by muscle weakness—may involve central fatigue mechanisms). These mild persistent symptoms are frustrating but don’t necessarily indicate treatment failure if they don’t significantly limit function.
Several factors contribute to ongoing symptoms despite treatment. Medication limitations include cholinesterase inhibitors providing only partial symptom relief (they improve neuromuscular transmission but don’t normalize it when receptor numbers remain reduced), immunosuppression taking months to years for maximum benefit (steroids start working in weeks but reach maximum effect at 6-12 months; steroid-sparing agents like azathioprine take 6-18 months for full benefit), and medication side effects sometimes mimicking MG symptoms (prednisone can cause muscle weakness and fatigue; this is steroid myopathy). Disease variability shows some patients simply have more refractory disease not fully responsive to standard treatments, and antibody levels and disease activity can fluctuate over time.
What you should discuss with your doctor includes whether you’re on optimal medication doses—is there room to increase pyridostigmine (up to 120 mg every 4 hours if needed, though higher doses have more side effects)? Is your prednisone dose adequate (usually 0.5-1 mg/kg daily initially, gradually tapered once improvement occurs)? Are you on steroid-sparing agents (azathioprine, mycophenolate, etc.) allowing prednisone reduction? Has enough time passed for medications to reach full effect (especially if recently started)? Are there undertreated factors including inadequate sleep (worsens fatigue), depression or anxiety (very common in chronic illness, worsens perceived fatigue), thyroid dysfunction (common in autoimmune disease, causes fatigue), or anemia or vitamin deficiencies?
Consider whether your activity level and expectations are realistic—are you expecting to do everything you did before MG at the same pace? Pacing activities, planning rest periods, and adjusting expectations are often necessary. That doesn’t mean accepting severe limitations, but recognizing that some accommodation may be needed. Are symptoms affecting major life functions (work, relationships, essential activities)? If so, escalation of treatment is warranted. If symptoms are mild annoyances not preventing important activities, current treatment may be optimal and further escalation (with attendant side effect risks) may not be worth it.
Options to discuss include adding or switching steroid-sparing agents if not already on them (azathioprine, mycophenolate most common), considering IVIg or plasmapheresis if symptoms are significantly limiting function despite oral medications (these provide temporary boost; some patients do periodic IVIg every 4-6 weeks for sustained benefit), evaluating for thymectomy if not yet done and you meet criteria (under 65, generalized MG, AChR antibody positive), or exploring newer biologics like eculizumab for refractory cases (though this is reserved for severe disease given cost and risks).
The bottom line is that completely normal function without any symptoms is an unrealistic expectation for most MG patients, but significant improvement allowing good quality of life and near-normal function is achievable for the majority. “Doing well” by physician standards should align reasonably with your subjective experience. If there’s a major disconnect (your doctor says you’re doing well but you feel severely limited), advocate for yourself—explain specifically how symptoms are affecting your life and ask about treatment optimization. Some persistent mild symptoms may be the price of avoiding more aggressive (and risky) treatments, and that trade-off is worth discussing explicitly so you and your doctor are aligned on goals.
Q3: I have myasthenia gravis affecting my swallowing and breathing. What are the warning signs of myasthenic crisis, and what should I do if I think I’m going into crisis?
Having myasthenia gravis with bulbar (swallowing) and respiratory involvement puts you at higher risk for myasthenic crisis, so understanding warning signs and having an action plan is critically important. Myasthenic crisis is defined as respiratory failure requiring intubation and mechanical ventilation, occurring in 15-20% of MG patients at some point. The mortality rate with appropriate treatment is about 5%, but crisis is a medical emergency requiring immediate hospitalization. Warning signs of impending or developing crisis include respiratory symptoms such as increasing shortness of breath especially when lying flat (orthopnea), needing to sleep propped up or in a chair, difficulty taking deep breaths or sighing, weak cough and difficulty clearing secretions, rapid shallow breathing, speaking in shorter phrases because of breathlessness, and anxiety or agitation (from air hunger and CO2 retention). Bulbar symptoms worsen with increasing difficulty swallowing both solids and liquids, choking on food or liquids, nasal regurgitation (liquids coming out nose when swallowing), drooling and inability to manage secretions, muffled or nasal voice quality, and difficulty holding head up (neck weakness).
Generalized weakness rapidly worsens with sudden escalation of symptoms beyond baseline, severe fatigue making even minimal activities exhausting, and difficulty with activities that were previously manageable. Precipitating factors often precede crisis including infections especially respiratory infections (pneumonia, bronchitis, URI), though any infection can trigger crisis. Medication changes or non-compliance includes stopping immunosuppressants, recent steroid dose reductions, or certain medications that worsen MG. Surgical stress or recent procedures, emotional or physical stress, pregnancy or postpartum period, and sometimes no identifiable trigger occurs. What to do if you suspect crisis: this is a medical emergency—do not wait to see if symptoms improve. Go to the emergency room immediately or call 911 if severely short of breath. Do not drive yourself—risk of respiratory failure en route.
Bring your medication list including all MG medications with doses, a written summary of your MG history if available (diagnosis date, antibody status, treatments tried, history of crisis), and your neurologist’s contact information. Inform ER immediately that you have myasthenia gravis and are concerned about crisis—many ER physicians aren’t familiar with MG, so advocating for yourself is critical. Tests that should be done include pulmonary function testing, specifically vital capacity and negative inspiratory force (NIF) measuring respiratory muscle strength. Arterial blood gas checking oxygen and CO2 levels. Chest X-ray looking for pneumonia or other lung problems. If vital capacity is below 15-20 mL/kg or NIF is less negative than -30 cm H2O, you’re at high risk for respiratory failure and need ICU monitoring even if not yet in overt failure.
Treatment in the hospital includes ICU admission for close monitoring of respiratory status, possible elective intubation if respiratory function is borderline rather than waiting for emergency intubation when you’re in extremis, plasmapheresis or IVIg immediately (work faster than steroids for crisis), high-dose IV steroids (typically methylprednisolone 1 gram daily for 3-5 days then high-dose oral prednisone), treating precipitating factors (antibiotics for infections, stopping offending medications), and stopping or reducing cholinesterase inhibitors temporarily since in severe weakness it’s sometimes hard to distinguish myasthenic crisis (too little ACh effect) from cholinergic crisis (too much ACh from medication overdose). Most patients improve within 2-4 weeks and are weaned from the ventilator, though some require longer support.
Prevention strategies include aggressive treatment of infections with early antibiotics for respiratory infections, keeping extra oral antibiotics at home for quick initiation if you develop infection symptoms, avoiding known MG-worsening medications (aminoglycosides, fluoroquinolones, beta-blockers, magnesium, etc.), maintaining medication compliance never skipping doses of immunosuppressants, avoiding rapid steroid tapers—gradual reduction only, monitoring for early warning signs with home pulse oximetry if you have history of respiratory involvement, and some patients monitor home vital capacity with a peak flow meter or portable spirometer. Having a written crisis action plan that you and your family understand is valuable, including what symptoms warrant immediate ER visit, which hospital to go to (ideally one with neurologists familiar with MG), medications to bring, and your neurologist’s emergency contact number.
Discussing advance directives with your doctor and family about your wishes regarding intubation and mechanical ventilation is important. Most MG patients recover from crisis and are successfully weaned from the ventilator, but discussing wishes beforehand is wise. The key message is that respiratory symptoms in MG should never be ignored or “waited out”—early recognition and treatment prevent full crisis and reduce ICU time and complications. Trust your instincts—if your breathing feels wrong, go to the ER. Better a false alarm than delayed treatment for true crisis.
Q4: I’ve read that thymus removal (thymectomy) might help my myasthenia gravis. How do I know if I should have this surgery, what are the risks, and how much improvement can I realistically expect?
Thymectomy is an important consideration for many MG patients, though not everyone is a candidate. Understanding who benefits and what to expect helps in decision-making. Indications for thymectomy include thymoma (thymus tumor)—if you have thymoma on chest imaging, thymectomy is mandatory regardless of MG severity since thymomas can be locally invasive or malignant. Removing the tumor is standard cancer treatment. About 10-15% of MG patients have thymoma, more common in older patients. Non-thymomatous MG has clearer indications including generalized MG (not pure ocular) with AChR antibody positive disease, age 18-65 (some centers extend to age 70-75), disease duration less than 5-7 years (benefit decreases with longer disease duration, though surgery can still help), and adequate strength to tolerate surgery (not in active crisis or severe weakness without stabilization first). Relative contraindications include pure ocular MG (benefit is uncertain, generally not recommended), anti-MuSK MG (studies show less benefit from thymectomy compared to AChR-positive MG), seronegative MG (benefit less clear but sometimes considered), elderly patients over 65-70 (benefit decreases with age; risks increase), and severe comorbidities making surgery risky.
Evidence for benefit came from the MGTX trial (2016), a landmark randomized controlled trial that compared thymectomy plus prednisone versus prednisone alone in non-thymomatous generalized MG patients. At 3 years, the thymectomy group had better clinical outcomes (lower MG symptoms scores), required lower prednisone doses, and had lower average prednisone doses over 3 years. This proved thymectomy is beneficial beyond just treating thymoma. Expected outcomes show 15-20% of patients achieving complete stable remission (off all medications, symptom-free) years after surgery. Improvement takes 2-5 years typically. About 50-60% have significant improvement allowing medication reduction or discontinuation of some medications, though most still need some treatment. About 70-80% have at least some improvement in symptoms or medication needs. About 20-30% have minimal or no benefit from surgery. Benefits continue accruing for up to 5-10 years post-surgery, so patience is needed.
Surgical approaches include median sternotomy (traditional)—splitting the breastbone (sternum) vertically to access the thymus. Provides best visualization and most complete thymus removal. Larger incision and longer recovery (4-6 weeks to full recovery). Minimally invasive options are video-assisted thoracoscopic surgery (VATS)—several small incisions in chest wall, using camera and instruments to remove thymus. Less pain and faster recovery (2-3 weeks). Possible incomplete removal if thymus has ectopic tissue. Robotic-assisted thoracoscopic surgery is similar to VATS but using robotic instruments. Some surgeons prefer this for precision. Most expensive approach. Extended trans-sternal thymectomy removes thymus plus surrounding fat/tissue where ectopic thymic tissue might hide—most complete removal. Some surgeons prefer this for non-thymomatous MG.
Surgical risks include typical surgical risks such as bleeding, infection, and anesthetic complications (important—some anesthetics worsen MG; anesthesiologist must be experienced with MG patients). Specific MG-related risks include post-operative myasthenic crisis (surgery is a stress that can trigger crisis—about 10% risk). Respiratory complications are common given underlying respiratory muscle weakness and pain from chest surgery. Prolonged ventilation may be needed (5-10% require ventilation longer than expected). Long-term complications include vocal cord paralysis if recurrent laryngeal nerve injured (rare, about 1%), phrenic nerve injury causing diaphragm paralysis (rare), and chronic pain or numbness at incision sites. Overall serious complication rate is about 5-10%, and mortality risk is less than 1% at experienced centers.
Pre-operative optimization is critical for safety including stabilization with medications (not operated during crisis or severe exacerbation), plasmapheresis or IVIg 1-2 weeks before surgery to optimize strength, respiratory function testing to establish baseline, cardiac evaluation especially if on chronic steroids, and anesthesia planning with anesthesiologist experienced in MG. Post-operative care involves ICU monitoring initially, aggressive respiratory therapy (incentive spirometry, chest PT to prevent pneumonia), pain control (adequate pain relief allows deep breathing), and continuation of MG medications (don’t stop suddenly).
Decision factors include weighing expected benefit (70-80% have some improvement but only 15-20% achieve remission; improvement takes years), risks (5-10% complication rate including crisis risk), age and disease characteristics (younger patients with recent-onset AChR-positive generalized MG benefit most), current disease control (if already well-controlled on medications, less urgency; if requiring high medication doses with side effects, more appealing), and personal preferences (some patients prefer avoiding surgery; others prefer potential to reduce medications). What I’d recommend is discussing with your neurologist whether you meet criteria for thymectomy based on age, MG type, antibody status, and disease duration. Get chest imaging if not already done to evaluate for thymoma and thymic hyperplasia. If you’re a candidate, meet with thoracic surgeons experienced in thymectomy for MG—discuss surgical approach options, their experience, and specific risks in your case. Consider seeking a second opinion at an MG center of excellence if uncertain. Understand that thymectomy is not a quick fix—benefits accrue slowly over years, medications continue post-surgery initially, and 20-30% may not benefit significantly. Many patients are glad they had surgery years later when they’re on lower medication doses or off medications entirely, even though the first few years post-surgery showed little obvious change.
Q5: I’m pregnant and have myasthenia gravis. How will pregnancy affect my MG, and is my baby at risk?
Pregnancy and myasthenia gravis interaction is complex and somewhat unpredictable, requiring careful monitoring and planning, but most women with MG can have successful pregnancies with good outcomes for both mother and baby. Your MG can change unpredictably during pregnancy—about one-third of women experience worsening of MG symptoms during pregnancy (most commonly in first trimester or postpartum), one-third experience improvement (possibly from hormonal or immunologic changes in pregnancy), and one-third stay stable with no significant change. Unfortunately, we cannot predict which pattern you’ll follow. Past pregnancies don’t necessarily predict future ones—you might improve in one pregnancy and worsen in another. Factors slightly increasing risk of worsening include prior history of exacerbations or crisis, bulbar or respiratory muscle involvement at baseline, and recent disease onset (within 1-2 years).
Management during pregnancy involves preconception counseling being ideal—optimize MG control before conception, review medications for pregnancy safety, establish baseline pulmonary function, and discuss risks and plans with neurologist and obstetrician. Medication considerations show pyridostigmine is safe in pregnancy (category B—no evidence of harm) and should be continued at the dose controlling symptoms. Prednisone is generally considered acceptable (category C—use if benefits outweigh risks). Corticosteroids in pregnancy, especially first trimester, slightly increase cleft palate risk, though absolute risk remains low. Benefits of MG control usually outweigh this small risk. Azathioprine is considered acceptable by many experts despite being pregnancy category D. Studies suggest less risk than previously thought. Discuss risks/benefits with your doctors. Mycophenolate mofetil is teratogenic—causes pregnancy loss and birth defects. Must be stopped at least 6 weeks before conception. Switch to alternative. Methotrexate is teratogenic—absolutely contraindicated. Must be stopped months before conception. Cyclosporine and tacrolimus have limited safety data but sometimes used when necessary—discuss with maternal-fetal medicine specialist.
Plasmapheresis and IVIg are safe in pregnancy if needed for severe exacerbations. Both can be used without harming the fetus. Monitoring during pregnancy includes neurology visits every 1-2 months throughout pregnancy with closer monitoring if symptoms worsen, pulmonary function testing each trimester and more frequently if respiratory symptoms develop, and early aggressive treatment of exacerbations (don’t wait—treat infections promptly, consider plasmapheresis or IVIg for moderate-severe worsening). Labor and delivery planning involves avoiding certain medications like magnesium sulfate (used for pre-eclampsia/eclampsia) which can precipitate myasthenic crisis. Must use alternative like calcium channel blockers. Alert obstetric team about MG and need to avoid magnesium. Epidural anesthesia is safe and preferred over general anesthesia, avoiding neuromuscular blocking agents (muscle relaxants used in general anesthesia) which can cause prolonged paralysis in MG patients or necessitating modified dosing if general anesthesia unavoidable.
Labor considerations include second stage of labor (pushing) potentially being exhausting for MG patients with severe muscle weakness. Some women require vacuum or forceps-assisted delivery. Cesarean section rates are slightly higher in MG patients but vaginal delivery is possible for most. Postpartum period is high-risk for MG exacerbation with about 30% of women experiencing worsening in first 6-8 weeks postpartum. Close monitoring is essential with arrangements for help at home. Newborn care can be exhausting—arrange for assistance. Breastfeeding is safe with most MG medications—pyridostigmine, prednisone, azathioprine, and IVIg pass into breast milk in small amounts but are generally considered safe. Discuss with pediatrician. Some women choose not to breastfeed given medication concerns and need for rest.
Neonatal myasthenia occurs in 10-20% of babies born to mothers with MG as maternal antibodies cross the placenta and temporarily affect the baby. Symptoms appear within first few days of life including weak cry, difficulty feeding, generalized hypotonia (“floppy baby”), and sometimes respiratory difficulties. The condition is temporary—as maternal antibodies clear from baby’s system over 2-4 weeks, symptoms resolve completely. Treatment is supportive with feeding support (may need NG tube temporarily), rarely needing pyridostigmine for severe symptoms, and sometimes requiring respiratory support (rarely mechanical ventilation). Almost all babies recover fully with no long-term effects. Importantly, the baby having neonatal MG does not mean they’ll develop MG later—this is temporary from maternal antibodies, not the baby’s own immune system. Future MG risk is slightly increased if mother has MG but remains low overall (about 1%).
Genetic considerations show MG is not directly inherited—it’s autoimmune, not a genetic disease passed parent-to-child like muscular dystrophies. However, there is a slightly increased risk of autoimmune diseases in children of parents with autoimmune disease (about 2-5% versus 1-2% in general population). Most children of MG mothers do not develop MG or any autoimmune disease. The bottom line is pregnancy with MG requires careful planning and monitoring but is usually successful. Most women have healthy pregnancies and healthy babies. The unpredictability of how MG will behave during pregnancy is challenging, but close monitoring and prompt treatment of exacerbations allow good outcomes. Working with neurologist familiar with MG in pregnancy and high-risk obstetrician (maternal-fetal medicine specialist) is ideal. Plan ahead for postpartum support given high risk of exacerbation and demands of newborn care. With appropriate care, women with MG can successfully have children.
Disclaimer
This article adapts publicly available information from medical databases and research organizations. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform — not a healthcare provider. Decisions about myasthenia gravis diagnosis, antibody testing, treatment with cholinesterase inhibitors, immunosuppression, thymectomy, or management of myasthenic crisis should be made in consultation with qualified physicians, neurologists, neuromuscular specialists, and multidisciplinary myasthenia gravis care teams who can evaluate your individual situation, antibody status, disease severity, and health circumstances. If you experience sudden worsening weakness, difficulty breathing, or swallowing problems, seek immediate emergency medical care.
References
- Myasthenia Gravis Foundation of America. About Myasthenia Gravis. https://myasthenia.org/
- Muscular Dystrophy Association. Myasthenia Gravis (MG). https://www.mda.org/disease/myasthenia-gravis
- PMC. Myasthenia Gravis: Clinical Features, Pathogenesis, and Treatment. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8289836/
- PMC. Myasthenia Gravis: Diagnosis and Management. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6822374/
- World Health Organization. Health Topics: Neuromuscular Disorders. https://www.who.int/health-topics/
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.