
Primary Sclerosing Cholangitis (PSC): Bile Duct Scarring and Its Link to IBD
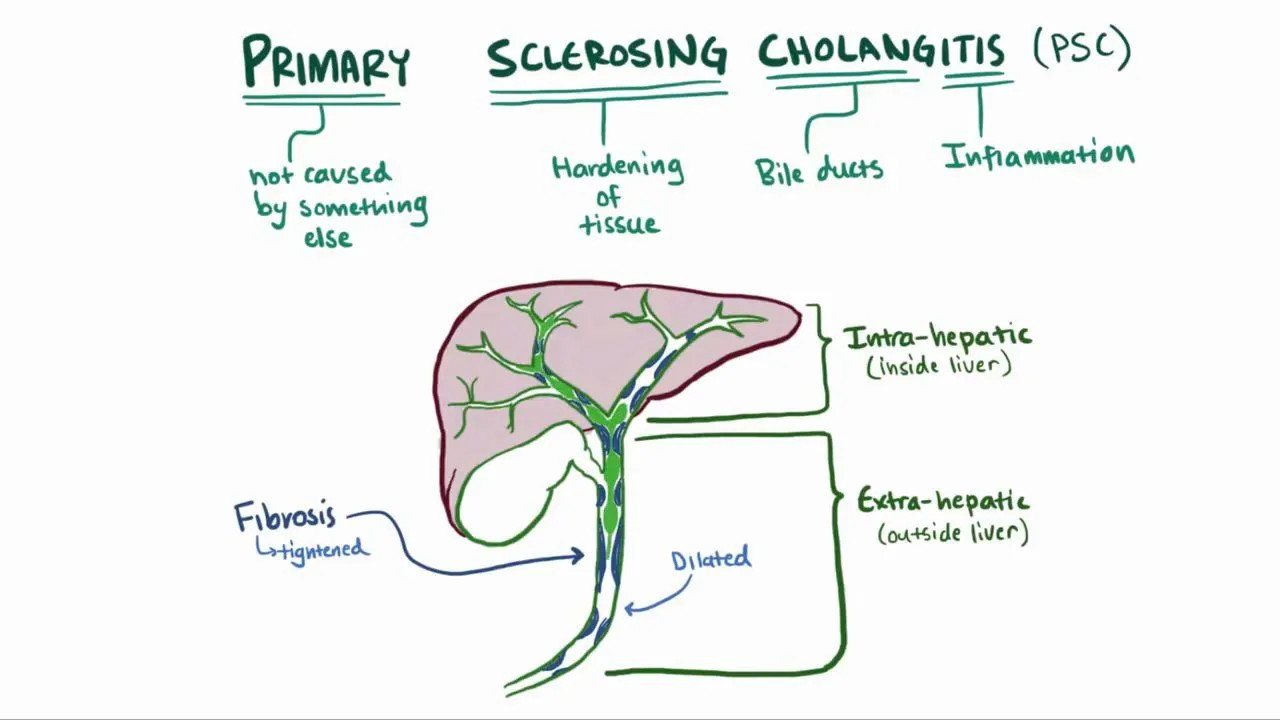
Imagine progressive scarring of bile ducts—the channels transporting digestive fluid through your liver—without obvious immune attack like in other autoimmune diseases. The ducts narrow from scarring. Bile backs up damaging your liver. Simultaneously, you suffer from inflammatory bowel disease causing chronic diarrhea and abdominal pain. Your liver and gut deteriorate together in this mysterious dual disease. This is primary sclerosing cholangitis—a rare, serious liver disease causing bile duct scarring, often occurring alongside inflammatory bowel disease. Primary sclerosing cholangitis, commonly abbreviated as PSC, is a chronic cholestatic liver disease characterized by progressive fibrosis and stricturing of bile ducts. Unlike primary biliary cholangitis where specific autoantibodies are found, PSC pathophysiology remains poorly understood. The disease causes progressive narrowing of bile ducts from scarring. Bile flow becomes impaired. Bile backs up causing cholestasis and liver damage. Progressive scarring leads to cirrhosis and liver failure if untreated. The most distinctive feature of PSC is its strong association with inflammatory bowel disease. Approximately 70 to 80 percent of PSC patients also have IBD—either ulcerative colitis or Crohn’s disease. Conversely, approximately 2 to 7 percent of IBD patients develop PSC. The reason for this strong association is incompletely understood but suggests shared pathophysiologic mechanisms. Primary sclerosing cholangitis affects approximately 1 to 2 per million people worldwide, though prevalence varies geographically. The disease is more common in Northern Europe and North America than in Southern Europe. Men are slightly more likely than women to develop PSC—approximately 60 percent of patients are men. This gender distribution differs from other autoimmune liver diseases which predominantly affect women. PSC typically develops in young to middle-aged adults, often between ages 30 and 50. What makes PSC particularly challenging is that no specific curative treatment exists. Disease progression is variable and unpredictable. Some patients progress slowly to cirrhosis over decades. Others progress rapidly requiring transplantation within years. Early diagnosis and treatment might slow progression but do not stop disease. In this comprehensive article, we will explore what primary sclerosing cholangitis is, understand the bile duct scarring process, recognize its distinctive link to inflammatory bowel disease, understand diagnosis methods, explore available treatments, and discover management strategies for this complex dual disease.
Understanding Bile Ducts and Sclerosis
Before we explore primary sclerosing cholangitis, we need to understand bile ducts and what happens during sclerosis—scarring. Bile ducts are tubular structures transporting bile from hepatocytes through the liver and into the intestine. Small intrahepatic ducts collect bile from liver tissue. These merge into progressively larger ducts. The largest duct within the liver is the hepatic duct. The hepatic duct merges with the cystic duct from the gallbladder forming the common bile duct. The common bile duct exits the liver entering the pancreas. The common bile duct empties into the small intestine (duodenum). Bile ducts are lined with cholangiocytes—specialized epithelial cells forming the bile duct lining. Cholangiocytes form a protective barrier between bile and the duct wall. Healthy bile ducts have smooth walls allowing normal bile flow. Bile flows continuously through the ducts into the intestine. Normal duct diameter is maintained by the underlying smooth muscle layer. The bile duct wall consists of multiple layers. The innermost lining is the epithelium composed of cholangiocytes. The submucosa is beneath the epithelium. The muscularis is the muscle layer. The serosa is the outer covering. Healthy bile ducts are flexible allowing changes in diameter with changing bile flow. Sclerosis is excessive scarring and fibrosis of tissues. In primary sclerosing cholangitis, scarring affects bile duct walls. The sclerosis process progressively thickens the duct walls. Fibrosis progressively reduces duct diameter. The narrowed ducts restrict bile flow. Bile becomes stagnant in narrowed areas. Stagnant bile causes further inflammation. The inflammation triggers more scarring. A vicious cycle of inflammation and scarring develops. Progressive narrowing eventually obstructs bile flow. Bile backs up into the liver. The accumulated bile damages hepatocytes. Hepatocytes die. The liver produces fibrosis attempting repair. Progressive liver fibrosis leads to cirrhosis. Understanding that sclerosis involves progressive narrowing of ducts explains why disease causes progressive cholestasis and eventually liver failure.
What is Primary Sclerosing Cholangitis?
Primary sclerosing cholangitis is a chronic cholestatic liver disease characterized by progressive inflammation, fibrosis, and stricturing of bile ducts. The disease causes progressive narrowing of bile ducts from excessive collagen deposition and scarring. The sclerosis affects both intrahepatic ducts (within the liver) and extrahepatic ducts (outside the liver) in different patterns. In primary sclerosing cholangitis, chronic inflammation of bile ducts leads to fibrosis. The inflammation is characterized by infiltration of lymphocytes and other immune cells. Inflammatory chemicals produced by immune cells trigger fibroblasts to produce excessive collagen. Collagen accumulates in bile duct walls. The accumulated collagen causes fibrosis and stricturing. Why the immune system attacks bile ducts in PSC is incompletely understood. Genetic factors are important—PSC runs in families. Specific HLA gene types increase susceptibility. However, genetics alone does not cause PSC. Environmental factors are also important. The strong association with IBD suggests that gut inflammation somehow triggers PSC development. Bacterial antigens from leaky gut might trigger immune response attacking bile ducts. Gut dysbiosis—abnormal bacterial composition—might contribute. Molecular mimicry—bacterial antigens resembling bile duct antigens—might activate immune cells attacking both bacteria and bile ducts. However, the exact mechanism remains unknown. PSC has three main patterns of bile duct involvement. Small-duct PSC affects only small intrahepatic ducts. The large ducts remain normal on imaging. Small-duct PSC represents approximately 10 to 15 percent of PSC cases. Small-duct PSC has better prognosis than large-duct PSC. Large-duct PSC affects the large intrahepatic and extrahepatic ducts. Stricturing of large ducts is visible on imaging. Large-duct PSC represents approximately 80 to 90 percent of PSC cases. Large-duct PSC has worse prognosis than small-duct disease. Overlap PSC occurs when PSC coexists with other autoimmune liver diseases. PSC-AIH (PSC with autoimmune hepatitis features) occurs in some patients. These overlap cases have features of both PSC and AIH. Overlap cases typically require treatment addressing both disease components. PSC is characterized by a distinctive appearance on imaging. The classic “beads-on-string” appearance shows alternating strictures and dilations. This beads-on-string pattern results from alternating narrowed and normal-appearing duct segments. The pattern is nearly pathognomonic for PSC. Progressive bile duct destruction is the hallmark. Without treatment, most PSC patients progress to cirrhosis over 10 to 20 years. However, disease progression is highly variable. Some patients remain stable for decades. Others progress rapidly to cirrhosis or require transplantation within years. Age at diagnosis influences prognosis. Younger patients generally have better prognosis than older patients. However, this might reflect longer observation time rather than slower disease progression.
The IBD Connection: Understanding the Bile Duct-Gut Link
The strong association between primary sclerosing cholangitis and inflammatory bowel disease is one of medicine’s most intriguing mysteries. Understanding this connection is crucial for comprehensive PSC management. Approximately 70 to 80 percent of PSC patients have inflammatory bowel disease. Most commonly, PSC is associated with ulcerative colitis. Approximately 60 percent of PSC patients with IBD have ulcerative colitis. Crohn’s disease occurs in approximately 30 percent of PSC patients with IBD. Some PSC patients have IBD features but do not meet diagnostic criteria for either UC or Crohn’s disease—they are classified as having IBD-unclassified. Conversely, approximately 2 to 7 percent of IBD patients develop PSC. IBD patients with PSC have worse prognosis than those without PSC. PSC increases colitis severity. PSC increases colon cancer risk in UC patients beyond the increased risk from UC alone. The dual disease causes significant morbidity. The reason for the strong PSC-IBD association remains incompletely understood despite decades of research. Genetic factors contribute. Patients with genetic predisposition to autoimmune diseases develop both PSC and IBD. Specific genetic markers are shared between PSC and IBD patients. Gut inflammation might trigger PSC development. In IBD, intestinal inflammation causes increased intestinal permeability—the leaky gut. Bacterial antigens from the gut lumen cross the damaged intestinal barrier. These bacterial antigens enter the bloodstream. The immune system becomes activated against these bacterial antigens. Through molecular mimicry, activated immune cells attack bile ducts expressing similar antigens. This bacterial antigen hypothesis explains why gut inflammation might trigger liver disease. Dysbiosis—abnormal bacterial composition in the gut—might contribute. PSC patients have altered gut bacterial composition compared to healthy controls. The dysbiosis might contribute to both IBD and PSC development. Molecular mimicry is the leading theory. Specific bacteria express antigens resembling bile duct antigens. Immune response against these bacteria cross-reacts attacking bile ducts. This molecular mimicry could explain simultaneous IBD and PSC development. However, proof of this theory remains elusive. Autoimmune mechanisms clearly overlap. PSC patients sometimes have autoantibodies. Antineutrophil cytoplasmic antibodies (ANCA) are found in some PSC patients. Anti-myeloperoxidase (MPO) ANCA is the most common pattern. These ANCA are also found in some IBD patients. The shared autoimmune markers suggest overlapping pathophysiology. Treatment implications are important. IBD treatment might influence PSC development. Some studies suggest 5-aminosalicylate drugs used for IBD might prevent PSC development. However, evidence is limited. Treating active IBD is crucial for PSC patients as IBD control might slow PSC progression. The PSC-IBD connection requires comprehensive management addressing both diseases.
Recognizing Early Symptoms: Signs of Bile Duct Scarring
Primary sclerosing cholangitis symptoms often develop insidiously, sometimes causing subtle symptoms initially attributed to other causes. Many early PSC patients are asymptomatic, discovered through abnormal liver tests. Recognizing symptoms when they develop prompts medical evaluation. Fatigue is an early and common symptom. Overwhelming exhaustion develops disproportionate to activity level. Fatigue affects daily functioning and quality of life. Fatigue sometimes precedes jaundice by months or years. Itching develops in many PSC patients. The itching results from accumulation of bile salts in blood. Bile salts deposit in skin causing intense itching. The itching is often worse at night disrupting sleep. Severe itching causes significant quality of life impairment. Jaundice develops as liver function deteriorates. Yellowing of skin and whites of eyes occurs. Jaundice results from elevated bilirubin in blood. Bilirubin accumulates because bile ducts cannot transport it normally. Jaundice indicates significant liver dysfunction. Abdominal pain or discomfort develops in some patients. The pain might be localized to upper right abdomen where the liver sits. The pain might be diffuse. Abdominal pain suggests hepatic inflammation or duct obstruction. Nausea and loss of appetite develop. Food becomes unappealing. Eating small amounts triggers nausea. These symptoms cause weight loss and malnutrition. Dark urine develops from elevated bilirubin. The urine becomes dark brown or cola-colored. Dark urine indicates elevated bilirubin in blood. Pale or clay-colored stools develop from reduced bile flow. Without bile reaching intestines, stool loses normal brown coloration. Pale stools indicate bile duct obstruction. Fever and chills sometimes develop. Fever suggests bacterial infection of obstructed bile ducts—cholangitis is a medical emergency. Fever with jaundice requires urgent evaluation and treatment. Cholangitis causes sepsis and can be life-threatening. Right upper quadrant pain develops in some patients. The pain is localized to the area over the liver. Sharp, severe pain suggests acute cholangitis or duct obstruction. Joint pain develops in some patients. Joints become painful and sometimes swollen. Joint pain sometimes precedes liver symptoms. Arthralgias might be attributed to arthritis before PSC is diagnosed. Many early PSC patients have no symptoms. The disease is discovered when blood tests ordered for other reasons show abnormal liver function. Asymptomatic patients discovered early generally have better outcomes than symptomatic patients. Some patients remain asymptomatic for years despite having PSC on imaging. Regular monitoring detects progression before symptoms develop.
Understanding Complications: Acute Cholangitis and Cholangiocarcinoma
Understanding serious complications of PSC helps recognize life-threatening situations requiring urgent treatment. Acute bacterial cholangitis is a medical emergency. Bacteria colonize obstructed bile ducts. The bacteria cause acute inflammation. Fever, severe pain, and jaundice develop acutely. Sepsis develops rapidly. Cholangitis causes systemic infection potentially fatal without urgent treatment. Cholangitis requires emergency treatment including antibiotics, fluid resuscitation, and biliary drainage. Recurrent cholangitis from repeated duct obstruction causes chronic morbidity. Cholangiocarcinoma—cancer of bile ducts—is one of the most serious complications. PSC patients have dramatically increased cholangiocarcinoma risk. Approximately 10 to 15 percent of PSC patients develop cholangiocarcinoma. The risk increases with disease duration. Cholangiocarcinoma typically develops in patients with advanced fibrosis. Cholangiocarcinoma is aggressive. Prognosis is poor—median survival is 1 to 2 years even with treatment. Screening for cholangiocarcinoma in PSC patients is challenging. No reliable screening test exists. Imaging surveillance is recommended but cannot reliably detect early cancer. By the time symptoms develop, cancer is typically advanced. Cholangiocarcinoma is the second leading cause of death in PSC patients after cirrhosis. Cirrhosis and portal hypertension develop as fibrosis progresses. Portal hypertension causes serious complications. Varices (enlarged veins in esophagus) develop. Varices can rupture causing life-threatening bleeding. Ascites (abdominal fluid) develops from portal hypertension. Hepatic encephalopathy develops from liver failure. Hepatorenal syndrome develops—kidney failure from severe liver disease. Kidney failure causes death without liver transplantation. Liver failure develops as cirrhosis becomes advanced. The liver cannot perform vital functions. Synthetic function fails—clotting factors decrease. Detoxification fails—ammonia accumulates. Albumin production decreases. Coma and death result without transplantation. Osteoporosis develops in PSC. Cholestasis impairs vitamin D absorption. Vitamin D deficiency causes calcium malabsorption. Liver disease impairs bone metabolism. Progressive bone loss increases fracture risk. Bone fractures from minor trauma indicate severe osteoporosis. These serious complications emphasize the importance of early diagnosis and treatment preventing or slowing disease progression.
Diagnosis: Recognizing PSC
Diagnosing primary sclerosing cholangitis requires combining clinical findings, liver tests, imaging, and sometimes cholangiography. Clinical history is crucial. Doctors ask about fatigue, jaundice, itching, and constitutional symptoms. They ask about IBD history. Family history of autoimmune disease is important. Physical examination assesses for jaundice, hepatomegaly, splenomegaly, and signs of cirrhosis. Blood tests are essential. Liver function tests assess cholestasis. Alkaline phosphatase (ALP) elevation is characteristic of PSC—often markedly elevated. Gamma-glutamyl transferase (GGT) elevation confirms hepatic origin of ALP. Bilirubin elevation develops as disease progresses. Transaminase (ALT, AST) elevation is mild in PSC. Albumin and prothrombin time (PT/INR) assess synthetic function. Low albumin and prolonged PT indicate advanced liver disease. Complete blood count sometimes shows cytopenias from portal hypertension. Autoantibody testing is performed. Unlike PBC, PSC patients typically lack anti-mitochondrial antibodies. ANCA antibodies (particularly anti-MPO ANCA) are found in some PSC patients. ANA is sometimes positive. The absence of specific autoantibodies makes PSC diagnosis more challenging than PBC. Imaging is crucial for PSC diagnosis. Magnetic resonance cholangiopancreatography (MRCP) is the diagnostic imaging of choice. MRCP shows the characteristic beads-on-string appearance of strictured ducts. MRCP reveals the pattern and extent of bile duct involvement. MRCP is non-invasive with no radiation or contrast injection. MRCP has largely replaced invasive ERCP for diagnosis. Ultrasound sometimes shows liver changes suggesting cirrhosis. CT can assess liver structure. Endoscopic retrograde cholangiopancreatography (ERCP) allows direct visualization of ducts. ERCP shows strictures and dilations. However, ERCP is invasive and carries risks including pancreatitis. ERCP is reserved for therapeutic interventions like stricture dilation or stent placement. Liver biopsy is sometimes performed for staging. Liver tissue shows periductal fibrosis—scarring around bile ducts. Fibrosis extends from ducts into surrounding liver tissue. The pattern of fibrosis distinguishes PSC from other cholestatic diseases. Biopsy findings help assess disease stage and prognosis. Colonoscopy should be performed in all PSC patients to assess for IBD. Even if patients deny IBD symptoms, subclinical IBD is common. Endoscopic findings guide IBD management. Diagnosis of PSC is confirmed when imaging (MRCP) shows characteristic bile duct changes (strictures and dilations) in appropriate clinical context. Liver biopsy confirms diagnosis if imaging is equivocal. Early diagnosis and staging guide treatment decisions and prognosis assessment.
Treatment: Managing Bile Duct Scarring and IBD
Primary sclerosing cholangitis treatment aims to slow disease progression, prevent complications, manage cholestasis, and treat coexisting IBD. Unfortunately, no specific disease-modifying therapy exists for PSC. Currently available treatments address symptoms and prevent complications rather than curing disease. Ursodeoxycholic acid (UDCA) is sometimes used. UDCA reduces cholestatic liver injury. However, evidence for UDCA benefit in PSC is limited unlike in PBC where UDCA is highly effective. Some studies show UDCA improves liver function tests. Other studies show no benefit. UDCA is generally considered safe but its role in PSC treatment remains uncertain. Obeticholic acid (OCA) is being studied for PSC. OCA is a farnesoid X receptor agonist. Early studies suggest OCA might slow PSC progression. However, OCA is not yet standard therapy for PSC. Fibrates including bezafibrate show promise. Fibrates reduce liver injury and improve liver function in some PSC patients. Bezafibrate might slow disease progression. However, evidence is limited and fibrates are not standard therapy. Antibiotics are used for acute cholangitis. Broad-spectrum antibiotics cover likely bacterial pathogens. Antibiotics prevent sepsis. Long-term prophylactic antibiotics are sometimes used in recurrent cholangitis. Biliary stricture management includes ERCP with stricture dilation and stent placement. ERCP allows therapeutic intervention. Balloon dilation widens strictured areas. Stent placement keeps strictures open. However, strictures frequently recur when stents are removed. Stricture management is palliative—it relieves obstruction but does not prevent disease progression. Immunosuppressive medications might help some patients. Corticosteroids, methotrexate, and azathioprine have been tried. However, evidence for immunosuppressive therapy benefit is limited. Some studies show no benefit from immunosuppression. Newer approaches targeting specific immune mechanisms are being studied. TNF inhibitors used for IBD might benefit PSC. Some studies suggest TNF inhibitors slow PSC progression. However, evidence is not conclusive. Treating coexisting IBD aggressively is important. 5-aminosalicylate drugs reduce inflammation. TNF inhibitors are effective in IBD. Adequate IBD control might slow PSC progression. Coordinating care with gastroenterologists ensures optimal IBD management. Liver transplantation is the only definitive treatment. Transplantation cures PSC by replacing the diseased liver. Transplanted liver functions normally for many years. Transplantation is indicated when cirrhosis develops or duct obstruction becomes unmanageable. PSC recurs in the transplanted liver in some patients requiring monitoring. Liver transplantation has excellent survival rates—5-year survival exceeds 80 percent.
Living with PSC: Daily Management and Monitoring
Living with primary sclerosing cholangitis requires regular medical monitoring, symptom management, treatment of coexisting IBD, and psychological adjustment. Attending hepatology appointments regularly ensures disease monitoring. Liver function tests assess disease activity. Imaging periodically assesses bile duct changes and fibrosis progression. Fibroscan monitors liver stiffness. Treatment adjustments are made based on disease progression. Regular monitoring detects complications early. Colonoscopy screens for IBD and colon cancer. PSC patients have increased colorectal cancer risk. Annual or biennial colonoscopy is recommended. Colonoscopy allows surveillance for dysplasia and cancer. Aggressively treating IBD is important. Active IBD causes ongoing gut inflammation. Gut inflammation might worsen PSC. Optimal IBD control might slow PSC progression. Coordinating care with gastroenterologists ensures both diseases are well managed. Itching management includes cholestyramine, rifampicin, or other agents. Various treatments must be tried to find what works. Multiple agents might be needed for severe itching. Avoiding triggers helps. Stress management reduces itching in some patients. Fatigue management includes activity pacing. Energy should be rationed to important activities. Rest periods are essential. Ensuring adequate sleep improves overall health. Work modifications might be necessary. Some patients require reduced work hours. Disability becomes necessary for some. Bone health protection is important. Calcium and vitamin D supplementation helps prevent osteoporosis. Weight-bearing exercise strengthens bones. Bone density monitoring guides treatment decisions. Bisphosphonates prevent bone loss if osteoporosis develops. Hepatotoxic medications must be avoided. Many medications damage liver. Acetaminophen at high doses damages liver. NSAIDs can impair liver function. Herbal supplements might damage liver. Medication use requires discussing with hepatologist. Alcohol must be avoided. Alcohol damages liver and accelerates disease progression. Complete alcohol avoidance is necessary. Vaccinations help prevent infections. Hepatitis A vaccine prevents hepatitis A. Hepatitis B vaccine prevents hepatitis B. Influenza vaccine prevents influenza. Pneumococcal vaccine prevents pneumococcal disease. Nutrition supports liver health. Adequate protein supports synthetic function. Balanced nutrition provides essential nutrients. Adequate hydration is important. Mental health support helps cope with chronic disease. Depression is common with chronic liver disease. Counseling addresses psychological effects. Support groups provide understanding from others with PSC. Antidepressants help some patients. Family and social support is invaluable. Educating loved ones about PSC helps understanding. Open communication about disease impact helps relationships navigate changes.
Frequently Asked Questions (FAQs)
Q1: Why is primary sclerosing cholangitis associated with inflammatory bowel disease?
The reason for the strong PSC-IBD association remains incompletely understood. Leading theories include: gut inflammation in IBD causing increased intestinal permeability allowing bacterial antigens to enter circulation; molecular mimicry where bacterial antigens resemble bile duct antigens triggering cross-reactive immune response; dysbiosis—abnormal bacterial composition—contributing to both diseases; and shared genetic predisposition to autoimmune diseases. The exact mechanism remains an area of active research.
Q2: Can primary sclerosing cholangitis be cured?
Currently, primary sclerosing cholangitis cannot be cured with medical therapy. Disease progression can sometimes be slowed with treatment but not stopped. Liver transplantation is the only definitive cure—it removes the diseased liver and halts PSC progression. However, PSC can recur in the transplanted liver in some patients. With transplantation, long-term survival is excellent.
Q3: Is every IBD patient at risk for developing PSC?
No, only approximately 2 to 7 percent of IBD patients develop PSC. Most IBD patients never develop PSC. However, IBD patients should be monitored for PSC. Any IBD patient with elevated alkaline phosphatase or jaundice should be evaluated for PSC. Genetic factors likely determine which IBD patients are at risk for PSC development.
Q4: How serious is the connection between PSC and colon cancer?
PSC significantly increases colon cancer risk in patients with ulcerative colitis. The cancer risk from the combination of PSC and UC is higher than from either disease alone. Annual or biennial colonoscopy with surveillance for dysplasia is recommended. Dysplasia found on colonoscopy indicates high cancer risk—colectomy might be recommended to prevent cancer development.
Q5: Can someone with PSC have a normal life expectancy?
Some PSC patients progress very slowly to cirrhosis over decades and might have normal life expectancy without developing severe disease. However, many PSC patients eventually develop cirrhosis or cholangiocarcinoma requiring transplantation. Life expectancy depends on disease progression rate and availability of liver transplantation. With modern treatment and timely transplantation, outcomes have improved significantly.
Key Takeaways
Primary sclerosing cholangitis is a chronic cholestatic liver disease causing progressive scarring and narrowing of bile ducts. Approximately 70 to 80 percent of PSC patients have inflammatory bowel disease—most commonly ulcerative colitis. The PSC-IBD association suggests shared pathophysiologic mechanisms though the exact link remains incompletely understood. PSC affects more men than women, differing from other autoimmune liver diseases. The characteristic beads-on-string appearance on MRCP imaging is nearly pathognomonic for PSC. Without specific autoantibodies, PSC diagnosis relies on imaging findings. Progressive bile duct scarring leads to cirrhosis and liver failure if untreated. Serious complications include acute bacterial cholangitis and cholangiocarcinoma. Currently, no specific disease-modifying therapy cures PSC. Liver transplantation is the only definitive treatment. Aggressive IBD management might slow PSC progression. With modern treatment and timely transplantation, outcomes have improved significantly. Regular monitoring detects complications early allowing intervention.
References
- World Health Organization (WHO). “Primary Sclerosing Cholangitis and Bile Duct Disease.” Retrieved from https://www.who.int/
- American College of Gastroenterology. “Primary Sclerosing Cholangitis: Clinical Guidelines.” Retrieved from https://gi.org/
- Mayo Clinic. “Primary Sclerosing Cholangitis: Causes and Treatment.” Retrieved from https://www.mayoclinic.org/
- Cleveland Clinic. “Primary Sclerosing Cholangitis: Complete Information.” Retrieved from https://my.clevelandclinic.org/
- American Liver Foundation. “Primary Sclerosing Cholangitis Patient Resources.” Retrieved from https://www.liverfoundation.org/
- National Institute of Diabetes and Digestive and Kidney Diseases. “Primary Sclerosing Cholangitis.” Retrieved from https://www.niddk.nih.gov/
Related Articles on ObserverVoice.com
Explore more health and science topics on our platform:
- Understanding Autoimmune Diseases: When Your Immune System Attacks Itself
- Inflammatory Bowel Disease (IBD): Crohn’s Disease vs Ulcerative Colitis
- The Liver: Function, Disease, and Maintaining Hepatic Health
- Primary Biliary Cholangitis (PBC): The Autoimmune Liver Bile Duct Disease
- Chronic Liver Disease: Understanding Progressive Cirrhosis
- Living with Chronic Illness: Adaptation and Support Strategies
Disclaimer
This article adapts publicly available information from WHO sources. This content is for informational and educational purposes only and does not constitute medical advice. [ObserverVoice.com] is a news and information platform — not a healthcare provider. If you suspect you have primary sclerosing cholangitis, experiencing fatigue, jaundice, or itching, consult a qualified hepatologist or gastroenterologist for proper evaluation. Early diagnosis is crucial for preventing cirrhosis and liver failure. Always seek guidance from licensed healthcare specialists for diagnosis and treatment.
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.
Follow Us on Twitter, Instagram, Facebook, & LinkedIn