
Pheochromocytoma: The Tumor That Causes Blood Pressure Spikes
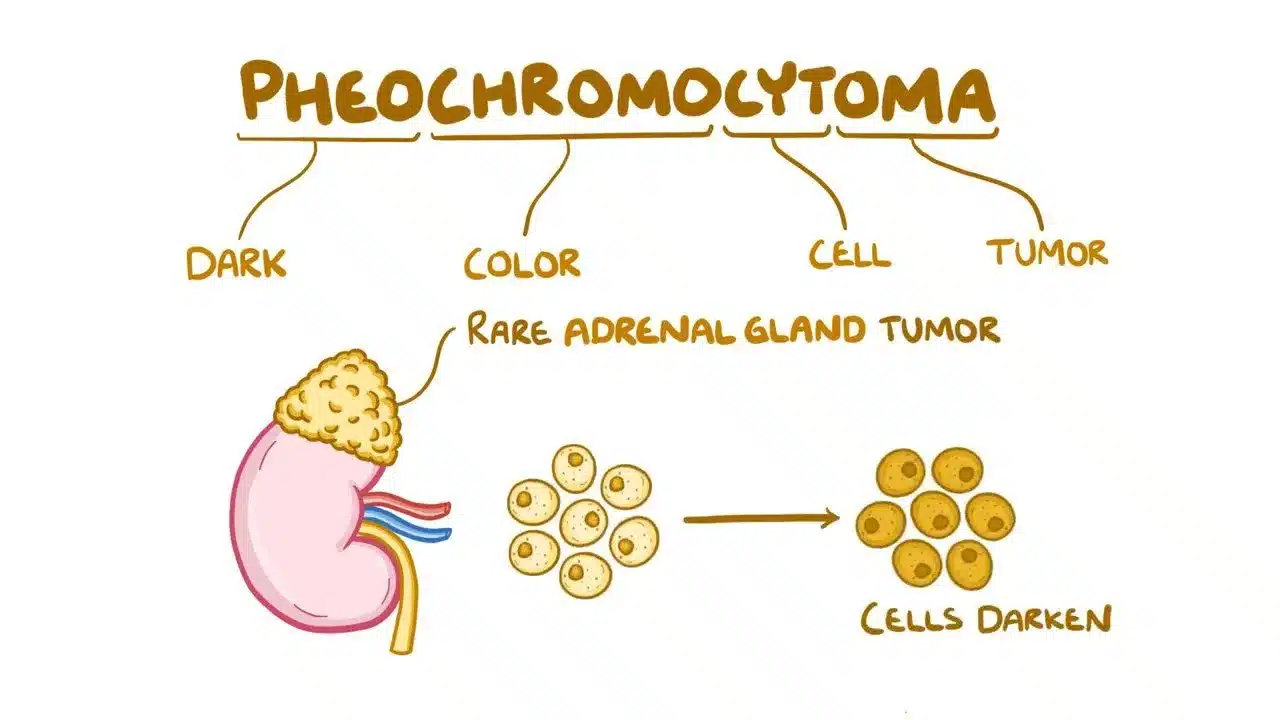
When 32-year-old Priya experienced sudden episodes of severe headache, heart pounding like a drum, and drenching sweats for no reason, her doctors initially thought she had panic attacks. But when her blood pressure soared to dangerous levels during these episodes—sometimes reaching 220/120 when normal is 120/80—further testing revealed a surprising cause: a small tumor in her adrenal gland producing massive amounts of adrenaline. This rare tumor, called pheochromocytoma, was literally flooding her body with stress hormones, causing her frightening symptoms. Pheochromocytoma affects only 2-8 people per million each year, but understanding this tumor is crucial because it’s one of the few causes of high blood pressure that can be completely cured with surgery.
What Are Adrenal Glands and What Does This Tumor Do?
Your adrenal glands are two small, triangular organs sitting on top of your kidneys like tiny hats. Each gland has two parts working like separate factories. The outer layer, called the cortex, produces hormones controlling salt balance, blood sugar, and stress response. The inner part, called the medulla, produces catecholamines—the body’s “fight or flight” hormones including adrenaline (epinephrine) and noradrenaline (norepinephrine). These hormones prepare your body for emergencies by increasing heart rate, raising blood pressure, and boosting alertness.
Pheochromocytoma is a tumor that grows in the adrenal medulla and acts like a hormone factory gone wild. Instead of producing adrenaline only when needed during stress or danger, the tumor releases massive amounts randomly throughout the day and night. Imagine your emergency alarm system constantly going off for no reason—that’s what happens inside the body with pheochromocytoma. Most of these tumors are benign, meaning they don’t spread to other organs, but about 10-15% are malignant and can spread to lymph nodes, bones, liver, or lungs.
These tumors vary in size from less than 1 centimeter (smaller than your fingernail) to over 10 centimeters (size of an orange), though most are 3-5 centimeters when discovered. About 90% occur in the adrenal glands, but 10% develop in similar hormone-producing tissue outside the adrenals along the spine, in the chest, or near the bladder. When these tumors occur outside the adrenal glands, doctors call them paragangliomas. The hormones released by the tumor—primarily norepinephrine and epinephrine—cause all the dramatic symptoms patients experience.
The Classic Symptoms: Sudden Attacks and Warning Signs
The hallmark of pheochromocytoma is the “spell” or “attack”—sudden episodes lasting minutes to hours when the tumor releases a burst of hormones. During these attacks, patients experience a characteristic trio of symptoms doctors call the “classic triad”: severe headache (pounding, throbbing pain often at the back of the head), profuse sweating (drenching sweats soaking through clothes, even in cool rooms), and rapid heartbeat (heart racing or pounding in the chest, sometimes feeling like it might jump out). These three symptoms together strongly suggest pheochromocytoma, especially when accompanied by very high blood pressure during attacks.
Other common symptoms include pale or flushed face (blood vessels constricting then dilating), tremors or shaking (like having too much coffee), chest pain or abdominal pain, nausea and vomiting, anxiety or sense of impending doom (feeling something terrible is about to happen), and weight loss despite normal appetite (high metabolism from constant adrenaline). The attacks can be triggered by various activities: physical exertion or exercise, bending over or changing position quickly, pressure on the abdomen (like tight clothing or massage), certain foods (aged cheese, wine, chocolate containing tyramine), certain medications (decongestants, stimulants), anesthesia or surgery (dangerous if tumor undiagnosed), and emotional stress or anxiety.
Between attacks, some patients feel completely normal while others have persistently elevated blood pressure. About 50% of patients have constant high blood pressure, 30% have episodic high blood pressure only during attacks, and 20% have normal blood pressure between episodes but dangerous spikes during attacks. This variability makes diagnosis challenging—many patients see multiple doctors over months or years before the tumor is discovered. In children, pheochromocytoma almost always causes sustained high blood pressure rather than episodic attacks.
How Doctors Diagnose This Sneaky Tumor
Diagnosing pheochromocytoma requires proving the tumor is producing excess hormones and then locating it with imaging. The first step involves measuring catecholamines and their breakdown products in urine or blood. The most reliable test is 24-hour urine collection measuring metanephrines and catecholamines—patients collect all urine over 24 hours in a special container, and the laboratory measures hormone levels. Elevated levels more than 2-3 times normal strongly suggest pheochromocytoma. Blood tests measuring plasma free metanephrines are also highly accurate and more convenient than 24-hour urine collection.
Before these tests, patients must avoid certain medications and foods that interfere with results: caffeine (coffee, tea, chocolate), acetaminophen (Tylenol), and certain blood pressure medications. Once biochemical tests confirm excess hormone production, imaging locates the tumor. CT scan or MRI of the abdomen and pelvis finds 95% of adrenal pheochromocytomas—the tumor appears as a bright mass on the adrenal gland. For the 10% of tumors outside the adrenal glands, a special nuclear medicine scan called MIBG scan uses radioactive tracer specifically taken up by catecholamine-producing cells, lighting up wherever the tumor hides in the body.
Genetic testing is recommended for all pheochromocytoma patients because 30-40% have hereditary syndromes, even without family history. Several genetic conditions increase pheochromocytoma risk. Multiple Endocrine Neoplasia type 2 (MEN2) causes pheochromocytomas plus thyroid cancer and parathyroid tumors. Von Hippel-Lindau disease (VHL) causes pheochromocytomas plus kidney cysts, pancreatic tumors, and brain/spine tumors. Neurofibromatosis type 1 (NF1) causes pheochromocytomas plus café-au-lait skin spots and nerve tumors. Hereditary paraganglioma syndromes (SDH mutations) cause pheochromocytomas and head-neck paragangliomas. Identifying these genetic mutations helps screen family members and guides surveillance for other tumors.
Treatment: Surgery Offers Cure for Most Patients
The definitive treatment for pheochromocytoma is surgical removal, which cures 95% of benign tumors. However, surgery is extremely dangerous without proper preparation because manipulating the tumor during removal can trigger massive hormone release causing life-threatening blood pressure spikes, heart rhythm problems, or stroke. Therefore, patients undergo 1-2 weeks of medical preparation before surgery. The first medication started is an alpha-blocker (phenoxybenzamine or doxazosin) blocking the effects of norepinephrine on blood vessels, preventing blood pressure spikes. Patients take increasing doses until symptoms controlled and blood pressure stable. Once alpha-blockade achieved, beta-blockers may be added controlling heart rate. Beta-blockers must never be given before alpha-blockers—this can cause dangerous blood pressure elevation.
During this preparation period, patients are instructed to consume high-salt diet and drink plenty of fluids expanding blood volume, preventing dangerous blood pressure drops after tumor removal. The surgery, called adrenalectomy, can be performed laparoscopically (minimally invasive with small incisions) for most adrenal tumors under 6-7 centimeters. Larger tumors or those with signs of malignancy require open surgery with larger incision. During surgery, the anesthesiologist monitors blood pressure and heart rate constantly, administering medications controlling extreme fluctuations as the surgeon manipulates and removes the tumor. Once the tumor is removed, blood pressure often drops suddenly—patients receive intravenous fluids and medications supporting blood pressure.
After successful surgery, catecholamine levels return to normal within days. Blood pressure normalizes in most patients, though some require continued blood pressure medications if they had long-standing hypertension causing permanent changes in blood vessels. Patients remain hospitalized 2-5 days monitoring blood pressure stability. Follow-up includes measuring urine or blood metanephrines 2-4 weeks after surgery confirming hormone levels normalized. If levels remain elevated, residual tumor tissue may be present requiring additional imaging and possible reoperation. For the 10-15% of malignant pheochromocytomas that spread, treatment is more complex involving surgery removing as much tumor as possible, radiation therapy targeting metastases, chemotherapy with cyclophosphamide, vincristine, and dacarbazine, and targeted radionuclide therapy using I-131 MIBG delivering radiation directly to tumor cells.
Living After Pheochromocytoma: Surveillance and Outlook
After successful surgery, most patients experience dramatic improvement—no more frightening attacks, normalized blood pressure, return to normal activities. However, lifelong surveillance is necessary because tumors can recur in 10-15% of patients and new tumors can develop, especially in hereditary cases. Surveillance involves measuring urine or blood metanephrines annually for at least 10 years detecting biochemical recurrence before symptoms develop. Imaging (CT or MRI) is performed if biochemical tests become abnormal or if new symptoms develop. For patients with hereditary syndromes, more frequent and comprehensive surveillance is needed screening for other associated tumors—thyroid ultrasound annually for MEN2, kidney and pancreas imaging for VHL, and full-body imaging for paraganglioma syndromes.
Pregnancy in women with pheochromocytoma history requires special management. Women planning pregnancy should have biochemical screening confirming no active tumor before conception because undiagnosed pheochromocytoma during pregnancy is extremely dangerous for both mother and baby. If pheochromocytoma discovered during pregnancy, surgery is ideally performed during second trimester when safest. If diagnosed late in pregnancy, medical management with alpha and beta-blockers continues until delivery, with tumor removal 4-6 weeks postpartum. Vaginal delivery avoided—cesarean section preferred preventing labor-induced catecholamine crisis.
Overall prognosis for benign pheochromocytoma is excellent—95% of patients cured with surgery, living normal lifespans with good quality of life. Even malignant pheochromocytoma has relatively favorable prognosis compared to other cancers—5-year survival rate 50-60% with treatment. The key to good outcomes is early diagnosis preventing complications from chronic catecholamine excess: heart muscle damage from constant adrenaline (cardiomyopathy), stroke from sudden blood pressure spikes, kidney damage from prolonged hypertension, and dangerous crisis during surgery or childbirth if undiagnosed.
Frequently Asked Questions
Q1: Can children get pheochromocytoma or is it only in adults?
Children can definitely develop pheochromocytoma, though it’s less common than in adults. About 10% of all pheochromocytomas occur in children, with peak age 9-12 years. In children, the tumor almost always causes sustained high blood pressure rather than episodic spells seen in adults. Children typically have severe headaches, vision problems from high blood pressure, excessive sweating, and poor weight gain despite good appetite. One important difference is that children have much higher rates of hereditary syndromes—up to 70-80% have genetic mutations compared to 30-40% in adults. The most common genetic causes in children are VHL disease, MEN2, and SDH mutations. This means all children diagnosed with pheochromocytoma should undergo comprehensive genetic testing, and their family members should be screened. Treatment is the same as adults—surgical removal after medical preparation with alpha and beta-blockers. Cure rates are equally excellent in children, and most grow and develop normally after successful treatment. However, children with hereditary syndromes require lifelong surveillance for tumor recurrence and other associated conditions.
Q2: What happens if someone with pheochromocytoma doesn’t know they have it and undergoes surgery for something else?
Undiagnosed pheochromocytoma during surgery is a medical emergency and can be fatal. When anesthesia is administered or the patient is intubated, the stress triggers massive catecholamine release from the tumor. This causes extreme blood pressure spikes (often over 300/200 mmHg), dangerous heart rhythms, heart attack, stroke, or pulmonary edema (fluid in lungs). Many cases of “unexplained” intraoperative crises are later discovered to be undiagnosed pheochromocytoma. This is why doctors screen high-risk patients before elective surgery—anyone with unexplained high blood pressure, episodic symptoms suggestive of catecholamine excess, or known genetic syndromes should have urine metanephrines checked before any surgery. If pheochromocytoma discovered, the planned surgery is postponed, the tumor is treated first with medical preparation and surgical removal, and then the original surgery can proceed safely 4-6 weeks later. Emergency surgery in patients with unsuspected pheochromocytoma requires aggressive medical management during the procedure—the anesthesiologist uses short-acting medications rapidly controlling blood pressure and heart rate fluctuations while the surgical team works quickly to complete the procedure.
Q3: If pheochromocytoma is removed successfully, can it come back?
Yes, pheochromocytoma can recur even after apparently complete removal. Recurrence rates are 10-15% overall but vary based on several factors. Malignant pheochromocytomas recur more frequently—30-50% develop recurrence or new metastases within 5-10 years. Hereditary pheochromocytomas also recur more often than sporadic cases—patients with VHL, SDH mutations, or MEN2 have higher risk of developing new tumors in the remaining adrenal gland or extra-adrenal locations. The timing of recurrence varies—some occur within 1-2 years suggesting incomplete initial resection or undetected second tumor, while others appear 10-20+ years later representing truly new tumor development. This is why lifelong surveillance is essential. Annual biochemical testing (urine or blood metanephrines) detects recurrence early before symptoms develop. If levels become elevated, imaging locates the recurrent tumor and repeat surgery is performed. Some patients undergo 3-4 surgeries over their lifetime removing recurrent tumors, especially those with hereditary syndromes. Despite recurrences, prognosis remains good if detected early through surveillance and treated promptly with surgery.
Q4: Are there any warning signs that a pheochromocytoma might be malignant?
Unfortunately, no reliable features distinguish benign from malignant pheochromocytoma at initial diagnosis. Unlike most cancers where pathologists can identify malignant cells under the microscope, benign and malignant pheochromocytomas look identical histologically. The only definitive way to diagnose malignancy is finding metastases—tumor spread to organs where chromaffin tissue normally doesn’t exist (lymph nodes, bones, liver, lungs). However, certain features suggest higher malignancy risk: larger tumor size (>5 centimeters), extra-adrenal location (paragangliomas have higher malignancy rates 15-35% versus 10% for adrenal tumors), certain genetic mutations (SDHB mutations associated 30-70% malignancy risk), very high dopamine levels (malignant tumors often secrete dopamine while benign ones typically don’t), and young age at diagnosis (<20 years). Even with these risk factors, many patients never develop metastases, and conversely, some seemingly low-risk tumors later prove malignant. This uncertainty is why all pheochromocytoma patients, regardless of apparent benign features, require long-term surveillance with annual biochemical testing and periodic imaging. Metastases can appear many years after initial diagnosis—sometimes 10-20+ years later—necessitating truly lifelong follow-up.
Q5: Can lifestyle changes or medications control pheochromocytoma without surgery?
Medical management alone is not curative and only used in specific situations when surgery impossible or delayed. Alpha and beta-blockers can control blood pressure and symptoms, making daily life more tolerable, but they don’t eliminate the tumor or prevent its growth. Long-term medical management without surgery carries significant risks: the tumor continues producing hormones causing progressive heart muscle damage (catecholamine cardiomyopathy), chronic high blood pressure damages kidneys, blood vessels, eyes, sudden massive hormone release can cause stroke, heart attack, or death, and for malignant tumors, delay allows metastatic spread reducing cure chances. Situations where medical management might be the only option include: very elderly patients (>80-85 years) with significant surgical risk and limited life expectancy, patients with widespread metastatic disease where surgery won’t cure but medications palliate symptoms, patients with severe medical conditions making any surgery extremely dangerous, and temporary management during pregnancy until delivery when surgery can be performed. Even in these cases, surgical removal remains the goal when patient’s condition improves or circumstances change. For everyone else, especially young healthy patients, surgery offers the only chance for cure and should be pursued after proper medical preparation. The dramatic improvement in quality of life after successful surgery—no more terrifying attacks, normalized blood pressure, freedom from multiple medications—makes surgery the clear treatment of choice when feasible.
Disclaimer
This article adapts publicly available information from publicly available medical databases and research organizations. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform — not a healthcare provider. Decisions about pheochromocytoma screening, diagnosis, and treatment should be made in consultation with qualified physicians, endocrinologists, and specialized surgical teams who can evaluate your individual symptoms, biochemical results, imaging findings, and health circumstances. If you experience sudden severe headaches, rapid heartbeat, or dangerous blood pressure spikes, please consult with your healthcare team immediately.
References
- National Cancer Institute. Pheochromocytoma and Paraganglioma Treatment. https://www.cancer.gov/types/pheochromocytoma/patient/pheochromocytoma-treatment-pdq
- Cleveland Clinic. Pheochromocytoma: Symptoms, Causes & Treatment. https://my.clevelandclinic.org/health/diseases/14325-pheochromocytoma
- Endocrine Society. Pheochromocytoma and Paraganglioma. https://www.endocrine.org/patient-engagement/endocrine-library/pheochromocytoma
- PMC. Pheochromocytoma: Clinical Manifestations, Diagnosis and Management. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6822347/
- World Health Organization. Cancer Topics. https://www.who.int/health-topics/cancer
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.