
Marfan Syndrome: The Connective Tissue Disorder Hidden in Tall, Thin Bodies
When 16-year-old Priya’s basketball coach noticed she was exceptionally tall and thin with unusually long arms and fingers, he encouraged her athletic potential. But during a routine physical exam, her doctor observed additional features—loose joints, a curved spine, and a heart murmur—prompting genetic testing that revealed Marfan syndrome, a connective tissue disorder affecting 1 in 5,000 people worldwide. Her doctor explained that while her height and build seemed like athletic advantages, they actually signaled a genetic condition affecting the “glue” holding her body together, with potentially life-threatening complications to her heart and blood vessels. Marfan syndrome is often called the “hidden disorder” because affected individuals may look remarkably healthy on the outside—tall, lean, and athletic—while facing serious internal complications including aortic aneurysms that can rupture without warning. Understanding Marfan syndrome is crucial because early diagnosis and treatment can prevent life-threatening complications, many affected individuals go undiagnosed until a medical crisis occurs, and with proper care, most patients live normal lifespans doing nearly everything their peers do.
Connective Tissue: The Body’s Framework That’s Built Incorrectly
Connective tissue is exactly what it sounds like—tissue that connects, supports, and holds together all the organs and structures in your body. It’s found everywhere: in your skin providing strength and elasticity, in blood vessel walls giving them flexibility to stretch with each heartbeat, in heart valves ensuring they open and close properly, in the lens of your eyes holding it in position, in bones providing a framework for calcium deposits, in joints allowing smooth movement, and in lungs providing elastic recoil for breathing. Connective tissue is composed of cells suspended in an extracellular matrix—a mesh-like network of proteins and other molecules the cells produce and secrete.
The most important protein in connective tissue is fibrillin-1, which forms long threadlike structures called microfibrils. These microfibrils act like scaffolding providing strength and elasticity to tissues. Think of fibrillin-1 like the steel framework inside a building—without it, the structure becomes weak and unstable. Microfibrils also regulate a growth factor called TGF-beta (transforming growth factor beta), keeping it inactive when bound to microfibrils. When microfibrils are defective, TGF-beta is released excessively, causing problems with tissue growth and remodeling.
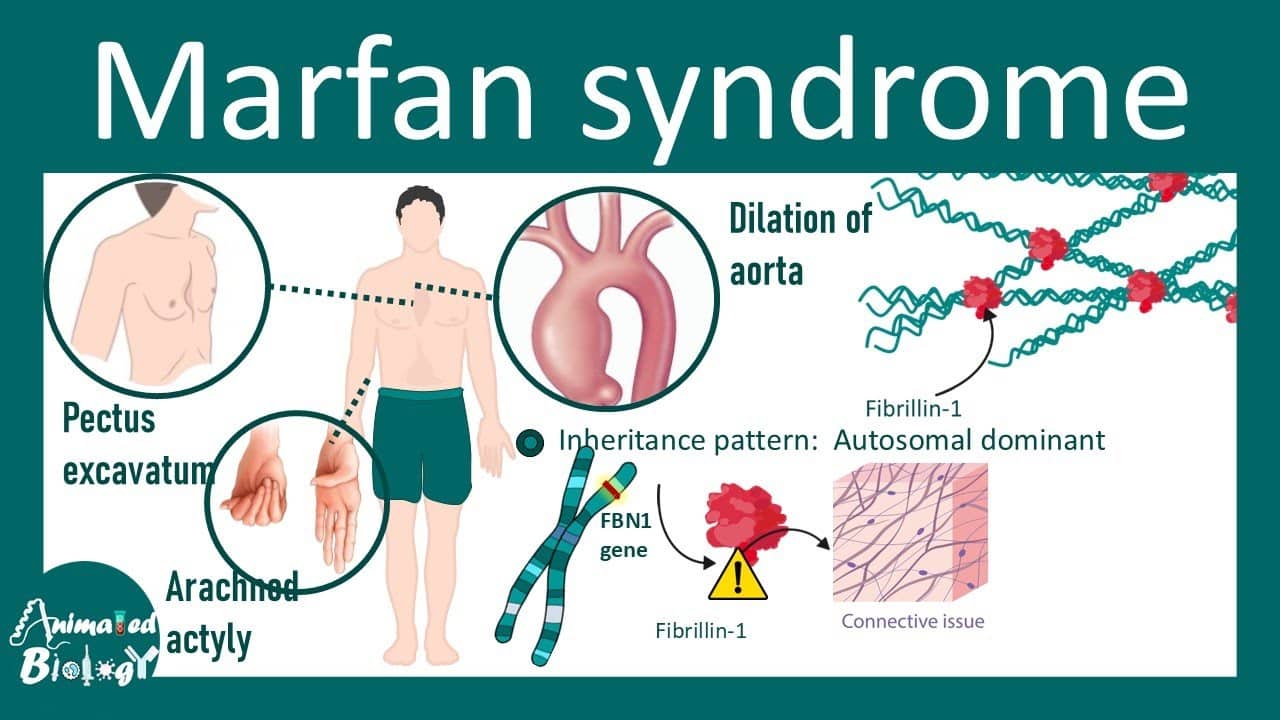
Marfan syndrome is caused by mutations in the FBN1 gene located on chromosome 15. This gene provides instructions for making fibrillin-1 protein. Over 3,000 different mutations in the FBN1 gene have been identified, each causing different amounts and types of defective fibrillin-1 production. Some mutations cause the gene to produce no fibrillin-1 at all, while others produce abnormal fibrillin-1 that doesn’t work properly. Without adequate normal fibrillin-1, microfibrils throughout the body are weak and unstable. Tissues that normally stretch and recoil—like blood vessels, heart valves, and ligaments—become overstretched, weakened, and prone to damage.
Marfan syndrome follows autosomal dominant inheritance, meaning only one copy of the mutated FBN1 gene (from one parent) is needed to cause the disorder. If a parent has Marfan syndrome, each child has a 50% chance of inheriting the mutation. However, 25-30% of cases result from new mutations occurring spontaneously during egg or sperm formation—these patients have no family history of Marfan syndrome, representing the first case in their family. The severity varies enormously even within the same family carrying the same mutation—some family members have mild features requiring minimal treatment, while others have severe complications needing multiple surgeries. This variability makes predicting disease course difficult.
Symptoms: A Body Built on Faulty Framework
Marfan syndrome affects multiple organ systems, creating a characteristic constellation of features though not every patient has every feature. Skeletal features are often the most visible. Tall stature with arm span exceeding height—when arms are stretched sideways, the measurement fingertip-to-fingertip is typically 5-10cm greater than the person’s height. Long, slender fingers and toes (arachnodactyly—literally “spider fingers”) create a distinctive appearance. The thumb sign (Steinberg sign) tests for this: when making a fist with thumb tucked inside, the thumb extends well beyond the edge of the hand. The wrist sign (Walker-Murdoch sign) occurs when wrapping thumb and pinky finger around the opposite wrist—they overlap significantly due to long fingers and narrow wrists.
Other skeletal features include pectus deformities where the chest either sinks inward (pectus excavatum—30-40% of patients) or protrudes outward (pectus carinatum—15-20%), affecting appearance and sometimes lung/heart function. Scoliosis (sideways spine curvature) develops in 50-60% of patients, ranging from mild requiring monitoring to severe needing surgery. Joint hypermobility makes joints excessively flexible—patients can bend joints beyond normal range, do contortionist-like positions, but suffer frequent dislocations, sprains, and chronic joint pain. Flat feet occur in most patients. The face may show distinctive features including long, narrow face, deep-set eyes, small jaw (retrognathia), and high-arched palate with crowded teeth.
Cardiovascular features are the most serious and life-threatening. Aortic root dilation occurs in 60-80% of patients—the aorta (main artery carrying blood from heart to body) gradually enlarges at its root where it exits the heart. Normal aortic root diameter is 2-3.5cm depending on body size; in Marfan syndrome it progressively dilates to 4, 5, 6cm or more. When it reaches 5-5.5cm, risk of aortic dissection (tear in the aortic wall) or rupture (bursting) becomes high—these are life-threatening emergencies requiring immediate surgery. Without treatment, aortic dissection or rupture causes sudden death in 30-40% of untreated Marfan patients, typically in their 30s-40s.
Mitral valve prolapse affects 60-90% of patients where the valve between left atrium and ventricle doesn’t close properly, allowing blood to leak backward (mitral regurgitation). Mild cases are asymptomatic; severe cases cause fatigue, shortness of breath, and heart failure. Tricuspid valve prolapse (20-30% of patients) involves the valve on the right side of the heart. Arrhythmias (irregular heartbeats) can develop. Heart failure occurs if valves leak severely or the aorta dilates excessively.
Eye problems affect 50-80% of patients. Ectopia lentis (lens dislocation) where the lens moves out of its normal position, usually upward and outward, occurs in 50-60% and is highly specific for Marfan syndrome. This causes blurred vision, double vision, or need for corrective lenses. Severe myopia (nearsightedness) develops early in childhood in 40-60% of patients. Retinal detachment risk is increased 10-fold compared to general population. Glaucoma and early cataracts can develop.
Lung problems occur less frequently but include spontaneous pneumothorax (collapsed lung) in 5-10% of patients—air leaks from lung into chest cavity, particularly in tall, thin patients. Obstructive sleep apnea develops in some patients. Stretch marks (striae) appear on skin not related to pregnancy or weight changes—found on shoulders, hips, lower back—from weakened connective tissue in skin. Dural ectasia affects 60-90% of patients—the dural sac surrounding the spinal cord and nerve roots in the lower back becomes enlarged, sometimes causing lower back pain, leg pain, numbness, or headaches, though many cases are asymptomatic discovered incidentally on imaging.
Diagnosis: Recognizing the Pattern
Diagnosing Marfan syndrome requires recognizing the pattern of features across multiple organ systems using the revised Ghent nosology criteria (updated 2010). These criteria assign points for various features, with diagnosis requiring a certain combination. In the absence of family history, diagnosis requires aortic root dilation AND ectopia lentis, or aortic root dilation AND FBN1 mutation, or aortic root dilation AND systemic features (skeletal, skin, dural ectasia) scoring ≥7 points. In patients with family history of Marfan syndrome, diagnosis requires ectopia lentis, or systemic features scoring ≥7 points, or aortic root dilation.
Cardiovascular evaluation is essential. Echocardiography (ultrasound of the heart) measures aortic root diameter, assesses valve function, and detects mitral/tricuspid prolapse. Measurements are compared to age and body-size adjusted norms—Z-scores calculate how many standard deviations above normal the aortic root is. Z-score ≥2 indicates dilation. CT or MRI angiography visualizes the entire aorta from heart through abdomen checking for aneurysms anywhere along its length, not just the root. Ophthalmology examination includes slit-lamp examination detecting lens dislocation, measurement of eye length (axial length—increased in Marfan), vision testing, intraocular pressure measurement (glaucoma screening), and dilated retinal examination (checking for detachment).
Genetic testing analyzes the FBN1 gene for mutations. Testing detects mutations in 90-95% of patients meeting clinical diagnostic criteria, confirming diagnosis and enabling testing of family members. However, 5-10% of patients clinically diagnosed with Marfan have no detectable FBN1 mutation despite having the disease—genetic testing limitations mean a negative test doesn’t rule out Marfan in someone with strong clinical features. Skeletal evaluation documents height, arm span, skeletal proportions, spine X-rays assessing scoliosis severity, chest imaging evaluating pectus deformities, and wrist/thumb signs.
Differential diagnosis—ruling out conditions that mimic Marfan—is important. Loeys-Dietz syndrome caused by mutations in TGF-beta receptor genes causes aortic aneurysms, skeletal features similar to Marfan but with hypertelorism (widely spaced eyes), cleft palate, and arterial tortuosity. More aggressive than Marfan with earlier aneurysms. Ehlers-Danlos syndrome causes joint hypermobility and skin hyperextensibility but usually not aortic problems (except vascular type EDS). Homocystinuria causes similar skeletal features, lens dislocation, and aortic problems but includes intellectual disability and different inheritance pattern. MASS phenotype (Mitral valve, Aorta, Skin, Skeletal) has mild Marfan-like features but doesn’t meet full diagnostic criteria and has better prognosis.
Treatment: Preventing Life-Threatening Complications
Treatment focuses on preventing aortic dissection and managing other complications. No cure exists, but with proper care most patients live normal lifespans. Cardiovascular monitoring and management is the highest priority. Beta-blockers (propranolol, atenolol) or losartan (an ARB—angiotensin receptor blocker) slow aortic root enlargement by reducing blood pressure and the force of each heartbeat against the aortic wall. Studies show these medications slow aortic growth by 30-50% and delay or prevent the need for surgery. Started in childhood, even before significant aortic dilation appears, and continued lifelong.
Regular echocardiography monitors aortic size—annually if aorta normal, every 6 months if mildly dilated, every 3-4 months if approaching surgical threshold. If the aortic root reaches 4.5-5.0cm (depending on body size, growth rate, family history), prophylactic aortic root replacement surgery is recommended before dissection occurs. The procedure replaces the diseased aortic root with a synthetic graft, sometimes requiring valve replacement (mechanical or tissue valve) or valve-sparing techniques preserving the patient’s own valve. Surgery performed electively is much safer than emergency surgery for dissection—elective mortality <2% versus emergency mortality 20-30%. After successful surgery, patients require lifelong monitoring of remaining aorta (which can still dilate) and anticoagulation if mechanical valve implanted.
For valve problems, mild mitral or tricuspid regurgitation requires monitoring only. Severe regurgitation causing symptoms or heart enlargement requires surgical repair or replacement. Lifestyle modifications are crucial. Avoid high-intensity contact sports (football, rugby, basketball, martial arts) and isometric exercises (weightlifting, rock climbing) that cause sudden blood pressure spikes potentially triggering dissection. Safe activities include swimming, golf, bowling, doubles tennis, hiking, cycling at moderate intensity. Pregnancy requires special management—aortic dissection risk increases during pregnancy, especially third trimester and delivery. Pre-pregnancy counseling assesses aortic size; if <4.0cm, pregnancy is relatively safe with close monitoring (monthly echocardiograms). If >4.0-4.5cm, prophylactic surgery before pregnancy may be recommended. Delivery at specialized centers with cardiac surgery backup, often by cesarean section avoiding labor stress.
Eye care includes annual ophthalmology exams, corrective lenses for myopia, surgical correction of lens dislocation if vision significantly impaired, prompt treatment of retinal detachment, and glaucoma management. Orthopedic management addresses scoliosis—monitoring with X-rays every 6-12 months during growth years, bracing for curves 25-40 degrees, surgery for curves >40-50 degrees. Pectus deformities sometimes require surgical correction if causing breathing problems or psychological distress. Physical therapy strengthens muscles supporting hypermobile joints reducing injury risk. Joint protection strategies avoid hyperextension and repetitive stress. Pain management for chronic joint pain may require medications, bracing, or occasionally surgery for severe joint damage.
Living a Full Life with Marfan Syndrome
With modern care, most people with Marfan syndrome live into their 70s—nearly normal lifespan. Before effective treatment existed, average life expectancy was 32 years, with most dying from aortic dissection. Today, with beta-blockers, aortic monitoring, and timely surgery, survival has improved dramatically. Patients can work, have families, and participate in most activities with certain restrictions. Education and career choices are unlimited—many successful professionals, artists, musicians, and academics have Marfan syndrome. Historical figures possibly affected include Abraham Lincoln (debated), Paganini (famous violinist—extreme finger length aided his playing), and others.
Athletic participation requires individualized assessment. Competitive high-intensity sports are discouraged due to dissection risk, but recreational low-intensity sports are encouraged for fitness and wellbeing. Some Marfan patients have competed in professional sports after careful evaluation and management, though this is controversial. Family planning involves genetic counseling—each child has 50% inheritance risk. Preimplantation genetic diagnosis with IVF allows selecting embryos without the mutation. Prenatal testing via amniocentesis or chorionic villus sampling detects the mutation in pregnancy, allowing informed decisions.
Psychosocial support addresses anxiety about health complications, body image concerns (distinctive appearance, surgical scars), activity restrictions, and genetic guilt about passing the condition to children. Support groups connect families affected by Marfan syndrome, sharing experiences and coping strategies. The Marfan Foundation provides education, research support, and community. Annual comprehensive evaluations at specialized Marfan clinics ensure all systems are monitored appropriately and emerging problems are detected early. The medical team typically includes cardiology, genetics, ophthalmology, orthopedics, and primary care coordinating care.
Frequently Asked Questions
Q1: Can someone with Marfan syndrome play sports, and which ones are safe versus dangerous?
This is one of the most common questions from young Marfan patients and parents, and the answer requires balancing safety concerns against quality of life and fitness benefits. The primary concern is aortic dissection triggered by sudden blood pressure spikes or chest trauma, so sport recommendations depend on intensity and contact level. Definitely avoid high-intensity competitive sports including basketball, football, rugby, hockey, wrestling, boxing, weightlifting, track and field (sprinting, throwing events), rock climbing, scuba diving (pressure changes), and competitive swimming. These cause extreme cardiovascular stress or chest trauma risk. Safe and encouraged low-to-moderate intensity activities include recreational swimming (excellent low-impact fitness), golf, bowling, doubles tennis (not intense singles), hiking and nature walks, cycling at moderate pace, yoga (avoiding extreme stretching in hypermobile joints), tai chi, fishing, and recreational jogging (if aorta stable and doctor approves). Borderline activities requiring individual assessment include running (marathons probably too intense; recreational 5Ks might be okay), singles tennis (more intense than doubles), skiing (avoid high speed/jumping), and dance (ballet’s hyperextension may stress hypermobile joints). The decision should involve your cardiologist reviewing your specific situation—aortic size (if normal or mildly dilated with stable growth, more activities permitted; if significantly dilated, more restrictions), blood pressure control (well-controlled on medications improves safety), family history (relatives with dissections at young ages suggest more caution), and age (children and adolescents have lower risk than adults, allowing more participation during youth). Many cardiologists recommend exercise stress testing measuring blood pressure response to exertion—if blood pressure remains well-controlled during exercise, moderate-intensity activities are safer. The key is staying physically active within safe parameters rather than becoming completely sedentary, which has its own health risks. Regular moderate exercise improves cardiovascular health, helps control blood pressure and weight, supports mental health, and maintains fitness. Work with your medical team finding activities you enjoy that are safe for your specific situation.
Q2: If I have Marfan syndrome, what are the warning signs of aortic dissection I should watch for and get emergency help immediately?
Aortic dissection is a medical emergency requiring immediate treatment—minutes matter. Every Marfan patient and their family should know the warning signs and understand to call emergency services immediately if they occur. The classic presentation is sudden, severe chest pain described as sharp, tearing, or ripping sensation starting in the center of chest and radiating to the back between shoulder blades. This differs from heart attack pain, which is usually described as pressure, squeezing, or heaviness rather than sharp tearing. Other symptoms include sudden severe back pain or abdominal pain depending on dissection location, sudden shortness of breath or difficulty breathing, loss of consciousness or feeling faint, sudden weakness or paralysis on one side of the body (if dissection affects arteries to brain), sudden vision changes or loss of vision, difficulty speaking or swallowing, weak or absent pulse in arms or legs (compared between sides—dissection can block blood flow to limbs), and cold, pale, or bluish limbs. Not every dissection presents with classic tearing chest pain—some have atypical symptoms or even mild pain, which is why any sudden new severe pain, especially in chest, back, or abdomen, warrants emergency evaluation in Marfan patients. If you experience these symptoms, call emergency services (ambulance) immediately—do not drive yourself or wait to see if symptoms improve. Tell emergency responders you have Marfan syndrome. At the hospital, CT angiography or transesophageal echocardiography quickly diagnoses dissection. Treatment involves emergency surgery for Type A dissections (involving ascending aorta near heart) and medications to lower blood pressure for many Type B dissections (involving descending aorta), sometimes requiring surgery later. Survival depends on rapid treatment—untreated aortic dissection is fatal in 50% of patients within 48 hours. With prompt surgical treatment, survival is 70-90%. Prevention is far better than treatment—taking blood pressure medications as prescribed, attending regular monitoring appointments, and having prophylactic surgery when aortic size reaches threshold prevents most dissections.
Q3: My child was just diagnosed with Marfan syndrome at age 8. What does this mean for their future and can they live a normal life?
Receiving a Marfan diagnosis for your child is undoubtedly frightening, but the prognosis today is remarkably good with proper care. Most children with Marfan syndrome grow up to live full, healthy, productive lives well into their 70s—approaching normal life expectancy. Here’s what to expect and how to ensure the best outcomes. Immediate steps include baseline evaluations with cardiology (echocardiogram measuring aortic root, valve function), ophthalmology (checking for lens dislocation, myopia), genetics (genetic testing confirming diagnosis, counseling about inheritance), and orthopedics (assessing spine, chest, joints). Starting medications early—even before significant aortic dilation appears—slows aortic growth. Beta-blockers or losartan started in childhood and continued lifelong reduce dissection risk substantially. Regular monitoring every 6-12 months tracks aortic growth, valve function, spine curvature, and eye changes, allowing early intervention if problems develop. Most children with Marfan live completely normal childhoods with minor restrictions—they attend regular school, participate in most activities (avoiding only high-intensity competitive sports and contact sports), play with friends, and develop normally. Many excel academically, artistically, or musically. The main challenges are managing medical appointments, taking daily medications, activity restrictions (which can be frustrating for athletic children), and sometimes teasing about appearance (tall, thin, long fingers—but many children view these as positive features). As they grow, monitoring intensifies during puberty when rapid growth can accelerate aortic dilation. If scoliosis develops, bracing or surgery may be needed. Some children require pectus surgery if chest deformity is severe. Most reach adulthood with stable aortas and good heart function. If aortic dilation progresses, elective surgery in late teens or twenties replaces the aortic root before dissection risk becomes high. After successful surgery, life continues normally with monitoring of remaining aorta. Your child can pursue any education or career, have relationships, marry, and have children (with genetic counseling about 50% inheritance risk and options like preimplantation genetic diagnosis). Many adults with Marfan have successful careers, families, and fulfilling lives. The key is excellent medical care, medication adherence, appropriate activity choices, and regular monitoring. Connect with the Marfan Foundation and support groups where you’ll meet adults and children thriving with Marfan. Focus on what your child can do rather than restrictions, normalize medical care as part of life, and help them understand their condition age-appropriately. With your support and modern medicine, your child’s future is bright.
Q4: How is Marfan syndrome different from being just naturally tall and thin? Could my tall, thin friend actually have undiagnosed Marfan?
Great question highlighting an important point—most tall, thin people don’t have Marfan syndrome. Being tall and thin alone isn’t enough for diagnosis; Marfan requires a constellation of specific features across multiple organ systems. However, many Marfan cases go undiagnosed until adulthood or even until a catastrophic event like aortic dissection, so heightened awareness is important. Features distinguishing Marfan from normal tall stature include extreme height—typically above 95th percentile with arm span exceeding height by >5cm. Most tall people have proportional arm span. Arachnodactyly—extremely long, thin fingers and toes with positive thumb and wrist signs. Regular tall people have proportional hands. Skeletal features like pectus deformities, scoliosis, joint hypermobility, long narrow face. Most tall people don’t have these. Cardiovascular features—aortic root dilation, mitral valve prolapse. These don’t occur in normal tall individuals. Eye features—lens dislocation, severe myopia from young age. Not present in regular tall people. Family history—multiple relatives with similar features, early heart problems, or sudden death. Your tall, thin friend should be evaluated if they have several of these features: arm span significantly exceeding height, extremely long fingers (can wrap thumb and pinky completely around wrist with significant overlap), chest deformity or scoliosis, extremely flexible joints (can bend thumb to forearm, put palms flat on floor without bending knees, do contortionist positions), stretch marks not from weight changes, heart murmur, very nearsighted from young age, or family history of aneurysms or sudden cardiac death. A simple screening is calculating arm span to height ratio—if arm span exceeds height by more than 5cm, warrants evaluation. The systemic score in Ghent criteria assigns points for various skeletal and other features—reaching certain thresholds suggests Marfan. If you’re concerned about yourself or someone else, see a doctor who can perform screening examination, order echocardiogram checking aortic root size, and refer to genetics if Marfan is suspected. Remember, early diagnosis is life-saving—undiagnosed Marfan patients risk sudden aortic dissection without warning. Better to be evaluated and reassured than to miss the diagnosis.
Q5: Can Marfan syndrome be cured with gene therapy or will it always require lifelong management?
Currently, Marfan syndrome cannot be cured and requires lifelong management, but exciting research offers hope for future therapies that might correct the underlying defect or significantly improve outcomes. Current treatment is purely symptomatic—medications slow aortic growth but don’t fix the defective fibrillin-1, surgery replaces damaged aorta but doesn’t prevent problems elsewhere, and monitoring detects complications but doesn’t prevent the disease. The fundamental problem—defective FBN1 gene producing abnormal fibrillin-1—remains uncorrected. Gene therapy research is exploring several approaches: gene replacement therapy would deliver a correct copy of the FBN1 gene to cells throughout the body using viral vectors, enabling production of normal fibrillin-1 alongside the defective version. Challenges include delivering the gene to sufficient cells in relevant tissues (blood vessels, heart, eyes, bones), maintaining long-term gene expression, and avoiding immune responses to the viral vector. Currently in very early research stages, years away from human trials. Gene editing using CRISPR technology could potentially correct the mutation in the patient’s own cells, but similar delivery challenges exist plus concerns about off-target effects. Antisense oligonucleotides could silence the mutated FBN1 gene while allowing the normal copy to function (Marfan patients have one normal and one mutated copy). This approach has shown promise in other genetic diseases but isn’t yet tested in Marfan. Small molecule drugs targeting downstream effects—losartan reduces TGF-beta signaling, which is overactive in Marfan. Clinical trials showed losartan slows aortic growth similar to beta-blockers, possibly better in some patients. Newer TGF-beta blockers being developed might be even more effective. Researchers are also studying drugs that help cells produce more fibrillin-1 from the normal gene copy or that stabilize defective fibrillin-1 making it more functional. Stem cell therapy is theoretically possible—replacing defective cells with healthy stem cells producing normal fibrillin-1—but faces enormous practical challenges in a condition affecting cells throughout the entire body. While cure isn’t available now, research is advancing, and treatments keep improving. Patients diagnosed today can expect even better therapies emerging during their lifetimes. Meanwhile, current management with medications and timely surgery allows most patients to live normal lifespans, so while lifelong management is required, the burden is manageable and outcomes are excellent with proper care.
Disclaimer
This article adapts publicly available information from medical databases and research organizations. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform — not a healthcare provider. Decisions about Marfan syndrome diagnosis, genetic testing, and treatment should be made in consultation with qualified physicians, cardiologists, geneticists, ophthalmologists, and specialists in connective tissue disorders who can evaluate your individual symptoms, family history, and health circumstances. If you have features suggestive of Marfan syndrome or experience sudden chest or back pain, please consult with your healthcare team immediately.
References
- National Institute of Arthritis and Musculoskeletal and Skin Diseases. Marfan Syndrome. https://www.niams.nih.gov/health-topics/marfan-syndrome
- The Marfan Foundation. What is Marfan Syndrome? https://marfan.org/about/marfan
- PMC. Marfan Syndrome: A Clinical Update. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5726744/
- PMC. Diagnosis and Management of Marfan Syndrome. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3042704/
- World Health Organization. Congenital Disorders. https://www.who.int/health-topics/congenital-anomalies
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.