
Huntington’s Disease: Genetic, Progressive, and Still Without a Cure
When 38-year-old Rajesh started making involuntary jerky movements with his hands and face—initially so subtle his family thought he was fidgeting nervously—he dismissed it as stress from work. But over months, the movements became more pronounced, he struggled with balance while walking, and his wife noticed personality changes including irritability and depression unlike his usual cheerful nature. After his father died from the same mysterious symptoms 15 years earlier, genetic testing confirmed what Rajesh feared: Huntington’s disease, a devastating inherited brain disorder caused by a defective gene passed from parent to child. This rare condition affects only 3-7 people per 100,000, but for families carrying the mutation, it represents a 50% chance each child will inherit the disease. Understanding Huntington’s disease is crucial because it’s one of the few genetic conditions where a single gene mutation guarantees developing the disease, genetic testing can predict disease decades before symptoms appear, and while no cure exists yet, groundbreaking research offers hope for treatments that might slow or prevent this relentless brain degeneration.
The Brain and the HTT Gene: Understanding What Goes Wrong
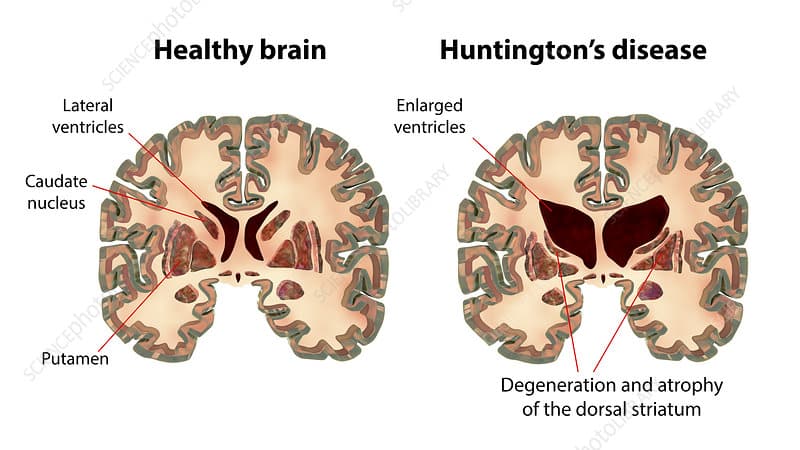
Your brain contains billions of nerve cells called neurons that communicate through chemical and electrical signals controlling every thought, movement, emotion, and memory. Deep inside your brain sits a group of structures called the basal ganglia—clusters of neurons responsible for coordinating movement, controlling muscle tone, and helping regulate emotions and thinking. The caudate nucleus and putamen (together called the striatum) within the basal ganglia are particularly important for smooth, coordinated voluntary movements and suppressing unwanted involuntary movements.
Huntington’s disease is caused by a mutation in the HTT gene (short for huntingtin) located on chromosome 4. This gene provides instructions for making a protein called huntingtin found throughout the body but especially concentrated in the brain. Normal huntingtin protein plays roles in nerve cell function, development, and survival, though scientists don’t fully understand all its functions. The mutation involves an abnormal repetition of three DNA building blocks—cytosine, adenine, and guanine (abbreviated CAG)—in the HTT gene. Normal people have 10-35 CAG repeats in their HTT gene, while people with Huntington’s disease have 36-120+ repeats.
This expanded CAG repeat causes the huntingtin protein to be produced with an abnormally long stretch of the amino acid glutamine (a CAG repeat codes for glutamine). The mutant huntingtin protein is toxic to neurons—it misfolds, clumps together forming aggregates inside neurons, and gradually kills brain cells, particularly in the striatum initially but eventually spreading throughout the brain. As neurons die, the brain physically shrinks—MRI scans show progressive brain atrophy with the caudate nucleus and putamen losing 50-95% of their volume over the disease course. The cerebral cortex (outer brain layer controlling thinking, personality, and judgment) also deteriorates causing cognitive and psychiatric symptoms.
The number of CAG repeats directly correlates with disease severity and age of onset. People with 36-39 repeats may not develop symptoms until age 60-70 or possibly never, while those with 40-50 repeats typically develop symptoms in their 30s-50s. Those with 60+ repeats can develop juvenile Huntington’s disease starting in childhood or adolescence—accounting for 5-10% of cases. Rarely, individuals have over 100 repeats causing symptoms in early childhood. The CAG repeat is unstable and can expand when passed from parent to child, particularly when inherited from the father—explaining why some children develop symptoms earlier and more severely than their affected parent, a phenomenon called anticipation.
Symptoms: Movement, Mind, and Mood All Affected
Huntington’s disease causes a triad of symptoms affecting movement (motor symptoms), thinking (cognitive symptoms), and emotions (psychiatric symptoms). The disease typically begins between ages 30-50, though onset ranges from childhood to age 80+. Early symptoms are often subtle and attributed to stress, aging, or other causes, delaying diagnosis for months to years. Motor symptoms are the most recognizable feature. Chorea—involuntary, rapid, irregular, dance-like movements—is the hallmark symptom. Initially affecting fingers, toes, or face (grimacing, tongue movements), chorea gradually spreads involving arms, legs, and trunk. Movements worsen with stress, improve with sleep, and cannot be voluntarily suppressed.
Other motor problems include dystonia (sustained muscle contractions causing twisting postures and repetitive movements), rigidity and stiffness especially in juvenile-onset cases, bradykinesia (slowness of movement), impaired balance and coordination causing frequent falls, difficulty walking with a lurching, unsteady gait, impaired eye movements (difficulty initiating voluntary eye movements, slow eye tracking), slurred speech (dysarthria) and swallowing difficulties (dysphagia) worsening as disease progresses. The swallowing problems become life-threatening—aspiration pneumonia (food/liquid entering lungs) is a common cause of death. Weight loss occurs despite adequate calorie intake due to the constant involuntary movements burning enormous energy.
Cognitive decline progresses from mild difficulties to severe dementia over 10-25 years. Early cognitive symptoms include difficulty organizing, planning, and prioritizing tasks (executive dysfunction), problems with multitasking and flexible thinking, slowed processing speed requiring more time to think and respond, memory problems especially retrieving information (though memory formation remains relatively intact initially), poor judgment and impaired insight, and difficulty learning new information. As disease advances, severe dementia develops with profound memory loss, inability to recognize family members, loss of speech, and complete dependence on caregivers.
Psychiatric and behavioral symptoms often appear before motor symptoms, sometimes 10-15 years earlier. Depression affects 40-50% of patients—persistent sadness, loss of interest in activities, sleep problems, hopelessness, and suicide risk (suicide rates are 4-6 times higher than general population, with highest risk around diagnosis time). Irritability and aggression develop causing anger outbursts disproportionate to triggers, argumentativeness, and sometimes physical aggression. Anxiety occurs with excessive worry, restlessness, and panic attacks. Apathy and loss of initiative cause reduced motivation, social withdrawal, and difficulty initiating activities. Obsessive-compulsive behaviors emerge with repetitive thoughts and actions. Psychosis can develop in 10-20% with paranoid delusions, hallucinations, or bizarre beliefs.
Juvenile Huntington’s disease (onset under age 20) presents differently than adult-onset. Motor symptoms include rigidity and stiffness more than chorea, slowness of movement (bradykinesia), tremor, and dystonia. Seizures occur in 30-50% of juvenile cases (rare in adult-onset). Rapid decline in school performance happens with learning difficulties, behavioral problems, and cognitive decline. The progression is generally faster than adult-onset, with death occurring 8-15 years after symptom onset.
Diagnosis: Genetic Testing Provides Definitive Answers
Diagnosing Huntington’s disease combines clinical evaluation, family history, brain imaging, and genetic testing. When symptoms suggest Huntington’s disease (involuntary movements, cognitive decline, psychiatric changes, family history), the neurologist performs detailed neurological examination checking for chorea, eye movement abnormalities, muscle tone, coordination, balance, reflexes, and cognitive function testing memory, processing speed, and executive function. Brain MRI typically shows atrophy (shrinkage) of the caudate nucleus and putamen causing enlargement of the fluid-filled spaces (ventricles) inside the brain. The degree of atrophy correlates with disease severity and duration.
Genetic testing provides definitive diagnosis by analyzing the HTT gene measuring the number of CAG repeats. A blood sample is sent to a specialized laboratory using polymerase chain reaction (PCR) to count the CAG repeats precisely. Results are interpreted as normal (10-35 repeats—will not develop Huntington’s disease), intermediate (36-39 repeats—may or may not develop symptoms, often in old age), and full penetrance (40+ repeats—will definitely develop Huntington’s disease if they live long enough). The test is 100% accurate with no false positives or false negatives. It can diagnose the disease decades before symptoms appear and can be performed prenatally if parents wish to know whether their unborn child carries the mutation.
Predictive testing—testing asymptomatic individuals at risk because they have an affected parent—is complex ethically and psychologically. About 10-20% of at-risk individuals choose predictive testing. The decision is deeply personal with profound implications. Benefits include ending uncertainty, allowing informed life planning (career, marriage, childbearing decisions), and enabling participation in clinical trials testing preventive therapies. Risks include psychological distress from positive results (depression, anxiety, suicide risk), genetic discrimination concerns regarding employment or insurance (though laws provide some protections), impact on family relationships, and survivor guilt if siblings test positive while they test negative.
Genetic counseling is mandatory before predictive testing, involving multiple sessions ensuring the person understands the test implications, is psychologically prepared, has adequate support systems, and is testing for their own reasons (not pressure from family members). Post-test counseling provides ongoing support regardless of results. For positive results, counselors help process emotions, connect with support resources, and plan for the future. For negative results, counselors address survivor guilt and adjustment to life without the disease cloud hanging overhead.
Preimplantation genetic diagnosis (PGD) allows at-risk parents to have biological children without passing on the mutation. In vitro fertilization creates embryos that are genetically tested before implantation, selecting only embryos without the HTT mutation for transfer to the uterus. This enables parents to have unaffected children without knowing their own genetic status if desired—embryos are tested and selected without revealing whether the at-risk parent carries the mutation. Some view this as eliminating Huntington’s disease from families; others have ethical concerns about embryo selection.
Treatment: Managing Symptoms and Maintaining Quality of Life
No cure exists for Huntington’s disease and no treatments slow the underlying brain degeneration. However, medications and supportive therapies significantly improve quality of life by managing symptoms. For chorea, several medications reduce involuntary movements. Tetrabenazine (Xenazine) depletes dopamine in the brain, reducing chorea in 70-80% of patients. Side effects include depression, sedation, and Parkinsonism (stiffness, slowness). Deutetrabenazine (Austedo) is a newer version with similar effectiveness but fewer side effects due to longer duration allowing twice-daily instead of three-times-daily dosing. Antipsychotic medications (haloperidol, risperidone) also reduce chorea by blocking dopamine receptors but carry risks of sedation, weight gain, and movement disorders.
For depression and anxiety, selective serotonin reuptake inhibitors (SSRIs) like sertraline, escitalopram, or fluoxetine effectively treat depression with relatively few side effects. Some patients require additional medications or psychotherapy. Close monitoring for suicide risk is essential especially around diagnosis time. For agitation, irritability, and aggression, mood stabilizers (valproic acid), atypical antipsychotics (quetiapine, olanzapine), or SSRIs help manage behavioral problems enabling patients to remain in home settings longer. For psychosis, antipsychotic medications (risperidone, olanzapine) treat delusions and hallucinations, though side effects require careful monitoring.
For insomnia, sleep hygiene measures (regular sleep schedule, cool dark bedroom, avoiding screens before bed) are tried first. If inadequate, medications like melatonin, trazodone, or zolpidem help. For juvenile Huntington’s with seizures, antiepileptic medications (levetiracetam, valproic acid) control seizures effectively. Physical therapy maintains mobility, flexibility, and balance for as long as possible, teaching safe walking techniques and fall prevention strategies. As disease progresses, therapists prescribe walkers, wheelchairs, and other adaptive equipment maintaining independence.
Occupational therapy helps maintain daily living skills (dressing, eating, bathing) through adaptive techniques and devices, modifies home environment for safety (removing tripping hazards, installing grab bars), and provides strategies for cognitive problems affecting daily function. Speech therapy addresses swallowing difficulties teaching safe swallowing techniques, recommending diet modifications (thickened liquids, pureed foods), and eventually advising feeding tube placement when swallowing becomes too dangerous. Therapists also help with communication difficulties as speech deteriorates, exploring alternative communication methods like writing boards or computer devices.
Nutritional support is crucial because patients burn 3,000-5,000+ calories daily from constant movements yet struggle eating due to chorea and swallowing problems. High-calorie diets with frequent snacks, nutritional supplements (protein shakes, smoothies), and eventually feeding tubes (gastrostomy tube placed through abdominal wall directly into stomach) maintain nutrition and hydration. Mental health support through psychologists, psychiatrists, and support groups helps patients and families cope with the emotional devastation of this disease. Hospice care in advanced stages focuses on comfort, dignity, and quality of life when cure is no longer possible.
Hope for the Future: Research Advances
While no cure exists today, research is advancing rapidly with several promising approaches in clinical trials. Gene silencing therapies aim to reduce production of the toxic mutant huntingtin protein using antisense oligonucleotides (ASOs)—synthetic molecules that bind to HTT messenger RNA preventing protein production. The drug tominersen showed promise in early trials reducing huntingtin protein levels in cerebrospinal fluid by 40-60%, but a phase 3 trial was discontinued in 2021 due to lack of benefit and possible harm—a disappointing setback but research continues with modified approaches.
Gene therapy attempts to deliver healthy HTT genes to brain cells or shut off the mutant gene. Challenges include getting therapies across the blood-brain barrier and into sufficient brain cells, but viral vector technologies are improving. Several gene therapies are in early development. Small molecule drugs targeting various aspects of disease are being tested including drugs that prevent huntingtin protein aggregation, enhance cellular waste disposal systems clearing toxic protein, protect mitochondria (cellular power plants damaged by mutant huntingtin), and reduce inflammation in the brain. Neuroprotective approaches like creatine, coenzyme Q10, and various vitamins have been tested but none have proven effective in large trials.
Stem cell therapies involve transplanting healthy neurons into the brain replacing those lost to disease. Early trials showed some neurons survive and integrate, but significant technical challenges remain. The Huntington Study Group, a network of research centers, conducts large observational studies following hundreds of patients tracking disease progression and identifying biomarkers predicting disease onset and progression. This data helps design better clinical trials. PREDICT-HD and TRACK-HD studies follow individuals with the gene mutation before symptom onset, trying to identify the earliest brain changes that might be targeted with preventive therapies.
Frequently Asked Questions
Q1: If my parent has Huntington’s disease, does that mean I will definitely get it too?
Not necessarily—you have a 50% chance of inheriting the mutant gene, meaning 50% chance of developing the disease and 50% chance of not inheriting it at all. Huntington’s disease follows autosomal dominant inheritance, meaning only one copy of the mutated gene (from one parent) is needed to develop the disease. When one parent has Huntington’s disease, each child has a 50/50 chance at conception of inheriting the mutated gene or the normal gene. If you inherit the normal gene, you will not develop Huntington’s disease and cannot pass it to your children—the disease stops with you. If you inherit the mutated gene with 40+ CAG repeats, you will develop the disease if you live long enough, though exactly when symptoms begin depends on the number of repeats. The only way to know your status is genetic testing. Many at-risk individuals choose not to test, preferring to live with uncertainty rather than knowing they will definitely develop this devastating disease. Others choose testing to end uncertainty and make informed life decisions. There is no right or wrong choice—it’s deeply personal. If you’re considering testing, genetic counseling is essential to help you think through the implications and prepare emotionally for results. Remember, even if you test positive, you may have 10-30 years before symptoms begin, and research is advancing rapidly with hope that treatments delaying onset or slowing progression may become available.
Q2: Can Huntington’s disease skip a generation or appear out of nowhere without family history?
Huntington’s disease never truly “skips a generation”—if someone carries the gene mutation, they will develop symptoms if they live long enough. However, it can appear to skip generations in certain situations. If a parent dies young (accident, other illness, suicide) before Huntington’s symptoms developed but carrying the gene unknowingly, they could pass it to children without the family knowing the disease existed. If a parent has 36-39 CAG repeats (reduced penetrance range), they might not develop symptoms until age 70-80+ or possibly never, but their child could inherit an expanded repeat (anticipation) causing symptoms much earlier, appearing like the disease “skipped” the parent. About 3-5% of Huntington’s cases have no known family history—called “apparently sporadic” cases. Some result from new mutations where the CAG repeat expanded from normal or intermediate range (27-35 repeats) to disease-causing range (40+) during egg or sperm formation. Others result from incorrect paternity, undocumented adoption, or family secrets where the disease existed but was hidden or misdiagnosed. Before genetic testing existed, many Huntington’s patients were misdiagnosed with other movement disorders, psychiatric illness, or dementia. Their descendants might not know they’re at risk. If you develop symptoms consistent with Huntington’s disease but have no family history, genetic testing can still diagnose the disease regardless of family history. About 1-3% of all cases represent true new mutations, though this is relatively rare.
Q3: What is the life expectancy after Huntington’s disease symptoms begin?
Life expectancy after symptom onset averages 10-30 years depending on age at onset and disease severity. Adult-onset Huntington’s (symptoms beginning ages 30-50) typically progresses over 15-20 years from diagnosis to death. The disease progresses through stages: early stage (5-8 years)—mild symptoms, independent living, working, driving. Middle stage (5-10 years)—increasing disability, requiring assistance with daily activities, unable to work or drive, significant chorea and cognitive decline. Late stage (3-5 years)—complete dependence, wheelchair or bed-bound, severe dementia, minimal communication, requiring total care. Death usually results from complications rather than the disease itself—aspiration pneumonia (60-70% of deaths) from food/liquid entering lungs due to swallowing difficulties, choking, falls causing fractures or head injuries, heart disease, suicide (especially early in disease), and infections. Juvenile-onset Huntington’s (symptoms before age 20) progresses more rapidly with death occurring 8-15 years after onset. Very late-onset cases (symptoms after age 60) often progress more slowly. Factors affecting survival include younger age at onset predicting longer disease duration, higher CAG repeat numbers associated with faster progression and shorter survival, presence of rigidity and bradykinesia (slowness) rather than chorea linked to faster decline, male gender associated with slightly shorter survival though reasons are unclear, and excellent supportive care (proper nutrition via feeding tubes, preventing pneumonia, treating infections aggressively) extending survival significantly. Some patients live 25-30 years with excellent care, while others decline more rapidly within 8-12 years.
Q4: Are there any lifestyle changes, diets, or supplements that can prevent or slow Huntington’s disease?
Unfortunately, no diet, supplement, or lifestyle modification has been proven to prevent symptom onset or slow progression in people carrying the HTT mutation. Many have been studied but none showed benefit in rigorous clinical trials. Tested but ineffective interventions include coenzyme Q10 (antioxidant supporting mitochondrial function), creatine (energy supplement), vitamin E (antioxidant), omega-3 fatty acids, curcumin, and various other supplements. Physical exercise shows promise in animal studies and small human trials suggesting it might have modest neuroprotective effects, but this remains unproven. That said, healthy lifestyle habits optimize overall health and wellbeing even if not altering disease course: regular exercise (aerobic, strength training, balance exercises) maintains cardiovascular fitness, muscle strength, coordination, and possibly cognitive function. Balanced nutrition with adequate calories maintains weight and energy. Brain-stimulating activities (reading, puzzles, social interaction) may support cognitive function. Stress management (meditation, yoga, counseling) supports mental health. Avoiding smoking and excessive alcohol protects cardiovascular and brain health. Treating coexisting conditions (diabetes, high blood pressure, depression) aggressively prevents additional brain damage. Adequate sleep supporting brain health and cellular repair processes. While these won’t prevent or cure Huntington’s, they optimize health allowing you to function at your best despite the disease. The most important thing is staying engaged in life, maintaining social connections, and accessing medical care and support services maximizing quality of life. Clinical trials are the best hope for finding treatments that actually slow progression—consider enrolling if eligible. Even if the experimental drug doesn’t help you, your participation advances research benefiting others.
Q5: Should I have children if I’m at risk for Huntington’s disease but haven’t been tested?
This is one of the most heart-wrenching decisions facing at-risk individuals, with no right or wrong answer—only what’s right for you. Some considerations: if you have children without testing, each child has a 25% chance of having Huntington’s disease (your 50% risk times their 50% chance if you’re positive). Some at-risk parents choose to have children accepting this risk, reasoning that life is worth living even with Huntington’s disease, and by the time their children develop symptoms, treatments or cures may exist. Others cannot accept the risk of passing this disease to children, choosing not to have biological children and instead adopting, remaining childless, or using assisted reproductive technologies. Options for having unaffected biological children include getting predictive testing—if you test negative, your children have no risk. If positive, you can consider other options below. Preimplantation genetic diagnosis (PGD) with IVF—embryos are created, tested, and only unaffected embryos implanted. Success rates are 40-50% per cycle. Cost is $15,000-25,000+ per attempt. This allows having unaffected children without learning your own genetic status if desired. Prenatal testing with chorionic villus sampling or amniocentesis tests the fetus during pregnancy. If affected, the pregnancy can be terminated if that aligns with your beliefs. Egg/sperm donation or embryo donation from unaffected donors ensures no Huntington’s risk. Adoption provides parenthood without genetic risk. Many at-risk individuals report that having children brought profound joy and meaning to their lives, even knowing they might later develop Huntington’s. Others feel relief choosing not to pass the gene on. There’s no “should”—only what you can live with. Genetic counselors, support groups, and mental health professionals can help you think through this decision, but ultimately only you can decide what’s right for your life circumstances, values, and beliefs.
Disclaimer
This article adapts publicly available information from medical databases and research organizations. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform — not a healthcare provider. Decisions about Huntington’s disease genetic testing, diagnosis, and treatment should be made in consultation with qualified physicians, neurologists, genetic counselors, and movement disorder specialists who can evaluate your individual symptoms, family history, and health circumstances. If you have involuntary movements, cognitive changes, or family history of Huntington’s disease, please consult with your healthcare team immediately.
References
- National Institute of Neurological Disorders and Stroke. Huntington’s Disease. https://www.ninds.nih.gov/health-information/disorders/huntingtons-disease
- Huntington’s Disease Society of America. What is HD? https://hdsa.org/what-is-hd/
- PMC. Huntington Disease: Natural History, Biomarkers and Prospects for Therapeutics. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4152532/
- PMC. Huntington’s Disease: Clinical Presentation, Pathogenesis and Treatment. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8071315/
- World Health Organization. Neurological Disorders. https://www.who.int/news-room/fact-sheets/detail/neurological-disorders
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.