
Amyotrophic Lateral Sclerosis (ALS): What Happens to the Body — and What Doesn’t
When 52-year-old Amit noticed his right hand becoming weak—struggling to button his shirt, dropping his coffee cup, and experiencing persistent muscle twitching in his thumb—he initially blamed aging or a pinched nerve. But when the weakness spread to his arm over several weeks and his speech became slurred, neurological testing revealed devastating news: amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease after the famous baseball player who died from it in 1941. This rare but relentless disease affects only 2-5 people per 100,000, yet it represents one of the cruelest diagnoses in medicine—progressively paralyzing every voluntary muscle in the body while leaving the mind completely intact. Understanding ALS is crucial because early recognition can improve quality of life through supportive care, new medications can modestly extend survival, and importantly, patients and families need to know what the disease affects and what it doesn’t—their thinking, personality, and awareness remain untouched even as their body fails around them.
Motor Neurons: The Communication Highway That Breaks Down
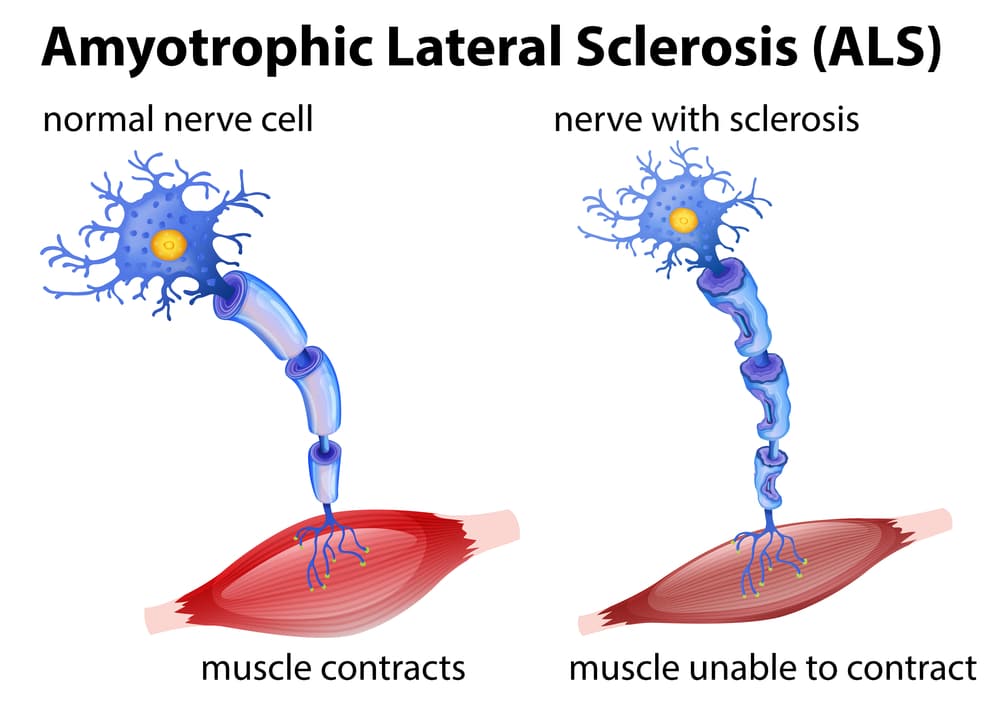
Your nervous system contains billions of nerve cells called neurons that transmit signals throughout your body controlling every function from breathing to thinking. Motor neurons are specialized nerve cells specifically controlling voluntary muscle movement—the muscles you consciously control like arms, legs, face, tongue, and breathing muscles. There are two types of motor neurons working together. Upper motor neurons originate in the motor cortex of your brain (the strip across the top of your brain controlling movement) and send signals down through your spinal cord. Lower motor neurons reside in the spinal cord and brainstem, receiving signals from upper motor neurons and transmitting them to muscles throughout the body.
When you decide to pick up a cup, upper motor neurons in your brain send electrical signals down through the spinal cord to lower motor neurons, which then transmit signals through long nerve fibers (axons) extending from the spinal cord to muscles in your arm and hand. At the junction between nerve and muscle (neuromuscular junction), the motor neuron releases a chemical messenger called acetylcholine causing muscle fibers to contract. This coordinated system allows smooth, precise voluntary movements. In ALS, both upper and lower motor neurons progressively degenerate and die for reasons scientists don’t fully understand. Without motor neuron signals, muscles cannot receive commands to move—they gradually weaken, shrink (atrophy), and eventually become completely paralyzed.
The name amyotrophic lateral sclerosis describes what happens: “amyotrophic” means muscle wasting (a=no, myo=muscle, trophic=nourishment), “lateral” refers to the sides of the spinal cord where affected nerve fibers travel, and “sclerosis” means scarring or hardening that occurs in damaged spinal cord areas. The critical point that distinguishes ALS from other neuromuscular diseases is that it affects only motor neurons—the nerve cells controlling movement. It does not affect sensory neurons (touch, pain, temperature sensation remain normal), autonomic neurons controlling automatic functions (heart rate, blood pressure, digestion, bladder and bowel control stay normal initially), and most importantly, it does not affect cognitive neurons in most cases (thinking, memory, personality remain intact in 95% of patients). This selectivity means ALS patients remain fully aware and mentally sharp even as their bodies progressively fail—a tragic aspect making this disease particularly devastating emotionally.
About 90-95% of ALS cases are sporadic, meaning they occur randomly without clear cause or family history. The remaining 5-10% are familial ALS, caused by inherited genetic mutations passed from parent to child. Over 25 genes associated with ALS have been identified. The most common are C9orf72 (40% of familial cases, also linked to frontotemporal dementia), SOD1 (20% of familial cases, was the first ALS gene discovered in 1993), TARDBP and FUS (each account for 4-5% of familial cases), and various others. Most familial ALS follows autosomal dominant inheritance—if a parent carries the mutation, each child has 50% chance of inheriting it. However, even with the mutation, not everyone develops ALS (incomplete penetrance), and age of onset varies. Risk factors for sporadic ALS include age (most common 55-75 years, rare before age 40), male gender (1.5 times more common in men), smoking (increases risk 1.5-2 fold), military service (veterans have higher rates for unclear reasons), exposure to heavy metals or pesticides (possible link but unproven), and intense physical activity or athletics (controversial association).
Symptoms: Progressive Weakness That Starts Somewhere, Then Spreads Everywhere
ALS symptoms begin subtly, often in one body region, then relentlessly spread to involve all voluntary muscles over months to years. There are three main patterns of onset. Limb-onset ALS (60-70% of cases) begins in arms or legs causing weakness in hands (difficulty buttoning, writing, using utensils, dropping objects), weakness in feet or legs (tripping, foot drop, difficulty climbing stairs), muscle cramps and stiffness especially at night, and visible muscle twitching (fasciculations) under the skin. The weakness typically starts asymmetrically—one hand or foot before the other—then gradually spreads to the opposite side and other limbs.
Bulbar-onset ALS (25-30% of cases) begins in muscles controlling speech and swallowing, causing slurred or nasal speech (dysarthria), difficulty swallowing liquids initially then solid foods (dysphagia), drooling from impaired saliva control, choking on food or liquids, and tongue weakness with visible fasciculations. Bulbar symptoms are particularly distressing, affecting communication and eating. Respiratory-onset ALS (rare, <5% of cases) begins with breathing muscle weakness causing shortness of breath especially when lying flat, morning headaches from carbon dioxide buildup overnight, poor sleep quality, and daytime fatigue. This is the least common initial presentation.
Regardless of onset pattern, ALS eventually affects all three regions—limbs, bulbar muscles, and breathing muscles. As the disease progresses, patients develop complete paralysis of legs and arms becoming wheelchair-bound then bed-bound, complete inability to speak (anarthria) though writing or computer communication possible until arm function lost, complete inability to swallow requiring feeding tube, and respiratory failure requiring mechanical ventilation or leading to death. The rate of progression varies enormously between patients. Some decline rapidly over 2-3 years from diagnosis to death, while others progress slowly over 10-15+ years. On average, 50% of patients survive 3 years from diagnosis, 20% survive 5 years, and 10% survive 10+ years. Factors predicting slower progression include limb-onset rather than bulbar-onset, younger age at diagnosis (<40 years), longer time between symptom onset and diagnosis, and absence of respiratory symptoms early in disease.
Critically, certain functions remain completely preserved in ALS. Sensation stays normal—patients feel touch, pain, temperature normally throughout the disease. Eye movements remain intact in most patients—even completely paralyzed patients can communicate using eye-tracking devices. Cognitive function stays intact in 95% of patients—thinking, memory, reasoning, personality all remain normal. About 5% develop frontotemporal dementia causing personality changes, behavioral problems, or language difficulties, but this is the exception. Bladder and bowel control remain normal until very late stages when immobility causes complications. Sexual function is not directly affected by ALS though physical limitations make intimacy challenging.
Diagnosis: Ruling Out Everything Else
Diagnosing ALS is challenging because no single test confirms it—diagnosis relies on recognizing the pattern of progressive upper and lower motor neuron damage while ruling out other conditions causing similar symptoms. The process typically takes 9-12 months from symptom onset to definitive diagnosis. The neurologist performs detailed examination checking for upper motor neuron signs including hyperreflexia (exaggerated reflexes), spasticity (muscle stiffness and increased tone), and Babinski sign (abnormal reflex response in the foot). Lower motor neuron signs include muscle weakness and atrophy, hyporeflexia (reduced or absent reflexes) in severely weak muscles, and fasciculations (visible muscle twitching).
The El Escorial criteria (revised in 1998) define ALS as progressive disease with both upper and lower motor neuron signs in multiple body regions, spreading from one region to others, with no other explanation for the symptoms. Electromyography (EMG) and nerve conduction studies are crucial tests. EMG involves inserting needle electrodes into muscles recording electrical activity, showing characteristic findings in ALS including fibrillation potentials and fasciculations at rest (indicating lower motor neuron damage) and large motor unit potentials (indicating surviving motor neurons taking over for dead ones). Nerve conduction studies measure how fast electrical signals travel along nerves—these are normal or only mildly affected in ALS, helping distinguish it from peripheral neuropathies.
MRI of brain and spine rules out other conditions mimicking ALS including cervical spondylotic myelopathy (spine degeneration compressing spinal cord), brain tumors, multiple sclerosis, and spinal cord tumors. Blood tests exclude treatable mimics like thyroid disease, vitamin B12 deficiency, heavy metal poisoning, Lyme disease, HIV, and paraneoplastic syndromes (neurological problems from hidden cancers). Lumbar puncture (spinal tap) analyzing cerebrospinal fluid may be done excluding infections or inflammatory conditions. Muscle biopsy is rarely needed but can help distinguish ALS from muscle diseases if diagnosis is uncertain.
Genetic testing is recommended for all ALS patients, not just those with family history, because 5-10% of apparently sporadic cases carry genetic mutations. Finding a mutation has implications for family members who may benefit from genetic counseling, presymptomatic testing, and eventually from gene-specific therapies being developed. The diagnosis profoundly impacts patients and families, so neurologists typically deliver the news carefully with family present, adequate time for questions, and immediate connection to ALS support resources including ALS Association, Muscular Dystrophy Association clinics, social workers, and support groups.
Treatment: Managing Symptoms and Maximizing Quality of Life
No cure exists for ALS, and treatments provide only modest benefits extending survival or slowing progression. However, comprehensive multidisciplinary care dramatically improves quality of life and may extend survival by many months. Riluzole (Rilutek) was the first FDA-approved medication for ALS in 1995. It modestly slows progression by reducing glutamate activity (an excitatory neurotransmitter potentially toxic to motor neurons at high levels). Studies show riluzole extends survival by 2-3 months on average. It’s given as 50mg twice daily orally. Side effects include elevated liver enzymes requiring monitoring, nausea, fatigue, and weakness. All ALS patients are candidates unless liver disease exists.
Edaravone (Radicava) approved in 2017, is an antioxidant reducing oxidative stress thought to contribute to motor neuron damage. Given intravenously in 28-day cycles (10 infusions over 14 days, then 14 days off). Studies showed it slowed functional decline in early-stage patients, though benefits are modest and not all patients respond. Side effects include bruising, gait disturbance, and headache. Oral edaravone (Radicava ORS) approved 2022 offers easier administration. Tofersen (Qalsody) approved in 2023 specifically for SOD1-mutated ALS (2% of all cases). It’s an antisense oligonucleotide reducing SOD1 protein production. Given as intrathecal injections (into spinal fluid) monthly. Early results show slowing of decline in SOD1 patients. This represents the first gene-specific ALS therapy.
Symptom management is crucial to quality of life. For muscle cramps and spasticity, baclofen, tizanidine, or gabapentin provide relief, while stretching and physical therapy maintain flexibility. For excessive saliva and drooling, anticholinergic medications (glycopyrrolate, amitriptyline) reduce saliva production, or botulinum toxin injections into salivary glands. For thick secretions, guaifenesin (expectorant) thins mucus and mechanical suction devices clear secretions. For pseudobulbar affect (uncontrollable laughing or crying inappropriate to emotions), dextromethorphan/quinidine (Nuedexta) effectively controls these episodes in 70-80% of patients. For depression and anxiety (affecting 40-50% of patients), SSRIs (sertraline, citalopram) and anxiolytics (lorazepam) combined with counseling provide support.
Respiratory support becomes necessary as breathing muscles weaken. Non-invasive ventilation (NIV) using BiPAP machines at night prolongs survival by 7-19 months and improves quality of life. Started when forced vital capacity drops below 50% predicted or symptomatic respiratory insufficiency develops. Invasive mechanical ventilation through tracheostomy maintains life indefinitely but requires 24-hour care and raises complex ethical issues. Only 5-10% of patients choose this option. Nutritional support prevents malnutrition from swallowing difficulties. High-calorie diets with soft foods are tried initially. Percutaneous endoscopic gastrostomy (PEG) tube—feeding tube placed through abdominal wall into stomach—is recommended when weight loss exceeds 10% or swallowing becomes unsafe. PEG tubes maintain nutrition, hydration, and medication administration, significantly improving quality of life. Ideally placed before respiratory function declines severely (FVC >50%) to reduce procedural risks.
Living with ALS: What Stays the Same, What Changes
Despite devastating physical limitations, many ALS patients find meaning and maintain quality of life through adaptive technologies, strong support systems, and focusing on what remains rather than what’s lost. Communication devices evolve as abilities change from standard speech, to writing when speech difficult, to computer-based communication using touch screens or keyboards, to eye-tracking devices when hands paralyzed, and finally to brain-computer interfaces in research settings. Modern augmentative communication devices allow patients to “speak” using synthesized voices, control their environment (lights, TV, phone), and access the internet and social media maintaining connections.
Mobility aids progress from canes and walkers initially, to manual then power wheelchairs, to specialized seating systems supporting paralyzed trunk and neck. Home modifications including ramps, widened doorways, stair lifts, and accessible bathrooms enable staying home longer. Personal care becomes necessary for bathing, dressing, toileting, and transfers. Many families provide care with home health aide support; others require skilled nursing facilities. Palliative care and hospice focus on comfort, dignity, and quality of life rather than life prolongation. Advance directives, living wills, and healthcare proxies established early ensure wishes regarding ventilation, resuscitation, and end-of-life care are followed.
Many ALS patients and families describe profound life transformations—priorities clarifying, relationships deepening, and appreciation for present moments intensifying. Support groups connect patients and families sharing experiences, coping strategies, and emotional support. The ALS Association and Muscular Dystrophy Association provide resources including equipment loans, support groups, educational materials, and advocacy. Research participation through clinical trials advances understanding and tests new therapies, giving patients hope they’re contributing to future treatments even if they don’t personally benefit.
Frequently Asked Questions
Q1: Is ALS always fatal, or can some people recover or live with it long-term?
ALS is currently always fatal—there are no documented cases of recovery or permanent remission. However, the timeline and disease course vary enormously between patients. The median survival from diagnosis is 2-5 years, meaning half of patients live longer than this and half less. About 20% survive 5+ years, 10% survive 10+ years, and 5% survive 20+ years from diagnosis. The most famous long-term survivor was physicist Stephen Hawking, who lived 55 years after his ALS diagnosis at age 21—extraordinarily rare but demonstrating the wide variability in disease progression. Factors associated with longer survival include younger age at onset (people diagnosed before age 40 tend to progress more slowly), limb-onset rather than bulbar-onset disease, longer time between first symptoms and diagnosis, early and aggressive use of riluzole and other treatments, excellent nutritional support with feeding tube when needed, early respiratory support with non-invasive ventilation, and comprehensive multidisciplinary care at ALS specialty clinics. Some patients choose invasive mechanical ventilation through tracheostomy, which can extend life indefinitely with proper care, though quality of life considerations make this choice uncommon (5-10% of patients). While ALS always ends in death from respiratory failure if ventilation is not used, maximizing the quality of years remaining through excellent supportive care, maintaining social connections, using adaptive technologies, and participating in meaningful activities allows many patients to live well with ALS for years.
Q2: Does ALS affect intelligence, memory, or personality, or does the mind stay completely normal?
This is one of the most important things to understand about ALS—in the vast majority of cases (95%), cognitive function, memory, and personality remain completely intact throughout the disease. Patients are fully aware, thinking clearly, remembering normally, and maintaining their personality and emotional capacity even as their body becomes completely paralyzed. This is both a blessing and a curse—blessing because they can continue relationships, make decisions, and find meaning in life; curse because they are fully aware of their progressive loss of function. However, 5% of ALS patients develop frontotemporal dementia (FTD), usually those with C9orf72 genetic mutations. FTD causes personality changes (becoming apathetic, socially inappropriate, or disinhibited), behavioral problems (compulsive behaviors, loss of empathy, poor judgment), language difficulties (trouble finding words, understanding speech, or speaking fluently), and executive dysfunction (problems with planning, organizing, problem-solving). When both ALS and FTD occur together, it’s sometimes called ALS-FTD. Importantly, even patients developing cognitive changes rarely lose awareness completely—they may have personality or language problems but usually maintain some level of awareness. Family members caring for ALS-FTD patients face additional challenges managing behavioral problems alongside physical disabilities. About 30-40% of ALS patients have very mild cognitive changes not meeting criteria for dementia—slight problems with multitasking, processing speed, or executive function detectable on testing but not significantly affecting daily life. The key message: assume all ALS patients have intact minds unless there’s clear evidence otherwise. Talk to them directly, not about them as if they’re not present. Include them in all decisions. Their paralyzed body houses a fully functioning mind.
Q3: Can ALS be prevented if it runs in my family, or can I find out if I’ll get it before symptoms start?
For the 5-10% of ALS cases that are familial (caused by inherited genetic mutations), genetic testing can determine if you carry the mutation before symptoms appear. If a parent had ALS and the specific gene mutation has been identified (like SOD1, C9orf72, etc.), children can undergo predictive testing to learn if they inherited the mutation. Testing involves genetic counseling explaining the implications, risks, and benefits, blood sample sent for genetic analysis of the specific mutation known in the family, and results revealing whether you carry the mutation (will likely develop ALS at some point) or don’t carry it (will not develop familial ALS, though still have general population’s very small risk of sporadic ALS). Currently, no proven prevention exists even if you test positive for a mutation. However, knowing has certain benefits: allows informed life planning (career, relationships, childbearing decisions), enables participation in clinical trials testing preventive therapies for mutation carriers, and gives time to prepare emotionally and practically. The decision to test is deeply personal—many at-risk individuals choose not to know, preferring to live without the knowledge they will definitely develop this disease. Others want to know to end uncertainty. For sporadic ALS (90-95% of cases), there’s no known prevention and no way to predict who will develop it. The only identified modifiable risk factor is smoking—quitting smoking reduces ALS risk. Proposed preventive measures like antioxidant vitamins, exercise, anti-inflammatory diets, etc., are unproven. Research is developing gene-specific therapies—for example, tofersen for SOD1 mutations—that might delay or prevent symptom onset if given to mutation carriers before symptoms appear. Clinical trials are testing this possibility. If your family carries a known ALS mutation, consider genetic counseling to discuss testing, regular neurological monitoring detecting earliest symptoms, and clinical trial opportunities.
Q4: What is the difference between ALS and other muscle-weakening diseases like muscular dystrophy or multiple sclerosis?
Though these conditions all cause weakness, they’re completely different diseases with different causes, symptoms, and outcomes. ALS affects motor neurons (nerve cells controlling muscles), causing muscle weakness because damaged nerves can’t send signals to muscles. The muscles themselves are normal initially—they weaken and shrink because they’re not receiving nerve signals. Sensation, vision, bladder/bowel control stay normal. Progression is relentless over 2-5 years typically. No treatments restore lost function. Fatal from respiratory failure. Muscular dystrophy is a group of genetic diseases affecting muscle tissue directly. The muscles themselves are defective, gradually breaking down and being replaced by fat and scar tissue. Nerves are normal—they send signals fine but muscles can’t respond. Different types exist (Duchenne, Becker, limb-girdle, etc.) with variable ages of onset and progression rates. Some forms are slowly progressive over decades; others advance rapidly. Treatment is supportive. Some types cause heart and breathing muscle problems leading to shortened lifespan. Multiple sclerosis (MS) is an autoimmune disease where the immune system attacks myelin (insulation around nerves) in the brain and spinal cord. Causes varied symptoms including vision problems, numbness/tingling, weakness, balance problems, bladder issues, fatigue, and cognitive changes. Symptoms often come and go in relapses and remissions rather than steady progression. Many patients have normal lifespans with treatment. Disease-modifying therapies slow progression and reduce relapses. The key differences: ALS affects motor neurons specifically, MS affects myelin in brain/spinal cord broadly, and muscular dystrophy affects muscles directly. ALS causes pure motor symptoms with preserved sensation, MS causes motor and sensory symptoms with relapses/remissions, muscular dystrophy causes progressive muscle weakness with some forms affecting heart. ALS progresses relentlessly and is always fatal within years, MS is usually not fatal and treatments can slow progression substantially, muscular dystrophy varies by type but many forms compatible with near-normal lifespan. If you’re diagnosed with one of these conditions, confirm which one because treatments and prognoses differ dramatically.
Q5: Should someone with ALS be put on a ventilator when breathing fails, and how do families make this decision?
This is one of the most difficult decisions ALS patients and families face, with no right or wrong answer. When breathing muscles weaken severely, two options exist: allow natural death from respiratory failure (death occurs peacefully usually within hours to days after stopping breathing, often with hospice providing comfort medications), or invasive mechanical ventilation through tracheostomy (surgically placed breathing tube in the neck connected to a ventilator machine breathing for the patient). Ventilation can extend life indefinitely—some ALS patients live 10-20+ years on ventilators. However, it requires 24-hour skilled nursing care, causes complete dependence, and raises quality of life questions. Considerations in the decision include quality of life with complete paralysis, dependence on machines, inability to speak (though communication possible with eye-tracking devices), and constant care needs. Some patients find life meaningful and valuable even completely paralyzed, maintaining relationships and activities through adaptive technology. Others consider this existence unacceptable. Financial and practical issues: 24-hour nursing care costs $200,000-400,000+ yearly. Many insurances provide limited coverage. Families may need to provide care or afford home health aides. Facility placement may be necessary but finding facilities accepting ventilator patients is challenging. Emotional impact on caregivers: providing 24-hour care for a ventilated patient is physically and emotionally exhausting. Marriages strain, families struggle, caregiver burnout is common. Patient preferences: some patients decide early they want “everything done” including ventilation; others are adamant about natural death without mechanical life support. The key is discussing preferences early before respiratory crisis occurs, documenting wishes in advance directives and medical orders, reviewing decisions periodically as disease progresses (people sometimes change their minds), involving family in discussions so everyone understands patient wishes. About 90-95% of ALS patients choose not to pursue invasive ventilation, opting for comfort care and natural death when breathing fails. The 5-10% choosing ventilation should be supported with excellent resources and care. Neither choice is wrong—it’s intensely personal based on individual values, beliefs, family situations, and resources.
Disclaimer
This article adapts publicly available information from medical databases and research organizations. This content is for informational and educational purposes only and does not constitute medical advice. ObserverVoice.com is a news and information platform — not a healthcare provider. Decisions about ALS diagnosis, genetic testing, and treatment should be made in consultation with qualified physicians, neurologists, ALS specialists, and multidisciplinary ALS clinics who can evaluate your individual symptoms, genetic risks, and health circumstances. If you experience progressive muscle weakness, speech changes, or swallowing difficulties, please consult with your healthcare team immediately.
References
- National Institute of Neurological Disorders and Stroke. Amyotrophic Lateral Sclerosis (ALS). https://www.ninds.nih.gov/health-information/disorders/amyotrophic-lateral-sclerosis-als
- ALS Association. What is ALS? https://www.als.org/understanding-als/what-is-als
- PMC. Amyotrophic Lateral Sclerosis: A Clinical Review. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7670675/
- PMC. ALS: Recent Advances in Pathogenesis and Treatment. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8234140/
- World Health Organization. Neurological Disorders. https://www.who.int/news-room/fact-sheets/detail/neurological-disorders
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.