
Nemaline Myopathy: Understanding a Rare Genetic Muscle Disorder
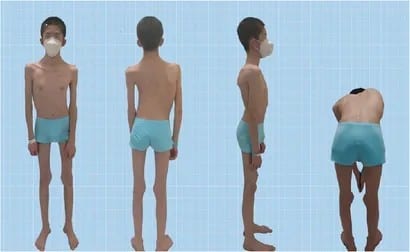
Nemaline myopathy represents one of the most extensively studied congenital myopathies, yet it remains a rare genetic disorder affecting approximately 1 in 50,000 individuals worldwide. First described in 1963 by Shy and colleagues, this condition derives its name from the Greek word “nema,” meaning thread, referring to the characteristic thread-like or rod-shaped structures found within affected muscle fibers. These microscopic abnormalities, known as nemaline bodies or rods, serve as the diagnostic hallmark of this complex neuromuscular disorder.
Pathophysiology and Molecular Mechanisms
The nemaline bodies that define this condition are electron-dense structures composed primarily of α-actinin and other sarcomeric proteins. These rods typically measure 1-7 micrometers in length and appear as purple-red inclusions when muscle tissue is stained with the Gomori trichrome technique. Under electron microscopy, they exhibit a characteristic lattice-like structure with a periodicity of approximately 180-200 nanometers.
The presence of these rods disrupts normal muscle fiber architecture and contractile function. While the exact mechanism by which nemaline bodies impair muscle contraction remains incompletely understood, research suggests they interfere with the sliding filament mechanism essential for muscle contraction by disrupting the organized arrangement of actin and myosin filaments.
Genetic Architecture
Nemaline myopathy exhibits remarkable genetic heterogeneity, with mutations identified in at least 13 different genes. The most commonly affected genes include:
- ACTA1 (Skeletal Muscle α-Actin): Accounting for approximately 15-25% of cases, mutations in this gene typically follow an autosomal dominant inheritance pattern and are associated with severe congenital forms of the disease.
- NEB (Nebulin): Responsible for approximately 50% of cases, nebulin mutations generally follow autosomal recessive inheritance. Nebulin is a giant protein that spans the thin filament and plays a crucial role in regulating actin filament length.
- TPM3 (Tropomyosin 3): Mutations in this gene account for about 2-3% of cases and can follow either autosomal dominant or recessive patterns. Tropomyosin is essential for regulating muscle contraction through its interaction with troponin.
- TNNT1 (Troponin T1): Found in approximately 3-5% of cases, these mutations typically follow autosomal recessive inheritance and are often associated with severe respiratory compromise.
Other genes implicated in nemaline myopathy include CFL2, KBTBD13, KLHL40, KLHL41, LMOD3, MYPN, RYR1, and TNNI2, each contributing to the disorder’s phenotypic diversity.
Clinical Manifestations
Phenotypic Spectrum
Nemaline myopathy presents with a broad clinical spectrum ranging from severe neonatal forms to mild adult-onset variants. This diversity reflects the underlying genetic heterogeneity and the variable impact of different mutations on muscle function.
- Severe Congenital Form: Affecting approximately 16% of patients, this variant manifests from birth with profound hypotonia, respiratory failure requiring ventilatory support, and severe feeding difficulties. Affected infants often present with arthrogryposis and may require intensive medical management throughout life.
- Intermediate Congenital Form: Representing about 20% of cases, this phenotype involves moderate weakness present from birth, delayed motor milestones, and variable respiratory involvement. Patients typically achieve independent ambulation but may experience progressive weakness over time.
- Typical Congenital Form: The most common presentation, accounting for approximately 46% of cases, involves mild to moderate weakness, delayed motor development, and characteristic facial features. Most patients achieve independent walking and have relatively stable muscle function.
- Mild Late-Onset Form: Comprising about 13% of cases, this variant may not be diagnosed until childhood, adolescence, or even adulthood. Patients typically present with gradually progressive weakness and may initially be misdiagnosed with other neuromuscular conditions.
- Adult-Onset Form: The rarest variant, representing less than 5% of cases, manifests in adulthood with slowly progressive weakness that may be initially attributed to other causes.
Clinical Features
The clinical presentation of nemaline myopathy is characterized by several distinctive features:
- Muscle Weakness: Predominantly affects proximal muscles, with particular involvement of neck flexors, facial muscles, and respiratory muscles. The weakness is typically non-progressive in congenital forms but may show slow progression in late-onset variants.
- Facial Dysmorphism: Patients often exhibit a characteristic elongated face with a high-arched palate, micrognathia, and ptosis. These features result from weakness of facial muscles and contribute to the distinctive appearance associated with the condition.
- Respiratory Compromise: Diaphragmatic and intercostal muscle weakness can lead to respiratory insufficiency, particularly during sleep. This complication represents one of the most significant clinical challenges in managing patients with nemaline myopathy.
- Skeletal Abnormalities: Progressive scoliosis, joint contractures, and foot deformities are common complications that may require orthopedic intervention.
Diagnostic Approaches
Histopathological Analysis
The gold standard for diagnosing nemaline myopathy remains muscle biopsy with specialized staining techniques. The Gomori trichrome stain reveals the characteristic purple-red nemaline rods within muscle fibers, while electron microscopy provides detailed visualization of their ultrastructural features.
Immunohistochemical studies using antibodies against specific proteins can help identify the underlying genetic defect. For example, absent or reduced α-actinin staining may suggest ACTA1 mutations, while abnormal nebulin distribution might indicate NEB gene involvement.
Genetic Testing
Comprehensive genetic testing has become increasingly important in diagnosing nemaline myopathy and providing accurate genetic counseling. Next-generation sequencing panels covering all known nemaline myopathy genes offer a high diagnostic yield, with molecular confirmation achieved in approximately 85-90% of cases.
Genetic testing not only confirms the diagnosis but also provides valuable information regarding inheritance patterns, prognosis, and potential targeted therapies. In families with known mutations, prenatal diagnosis and preimplantation genetic testing options are available.
Functional Assessments
Pulmonary function testing is crucial for evaluating respiratory muscle strength and identifying patients at risk for respiratory complications. Regular monitoring of forced vital capacity, maximum inspiratory pressure, and sleep studies helps guide respiratory management decisions.
Cardiac evaluation, including echocardiography and electrocardiography, is important as some patients may develop cardiomyopathy, particularly those with mutations in genes affecting cardiac muscle function.
Management and Treatment Strategies
Supportive Care
Current management of nemaline myopathy focuses on supportive care and symptom management, as no specific treatments are available to address the underlying genetic defects.
- Respiratory Support: Non-invasive ventilation, particularly during sleep, can significantly improve quality of life and survival in patients with respiratory muscle weakness. Some patients may require tracheostomy and mechanical ventilation for severe respiratory compromise.
- Physical Therapy: Regular physical therapy helps maintain joint mobility, prevent contractures, and preserve functional capacity. Aquatic therapy is particularly beneficial as it provides resistance training while supporting body weight.
- Orthopedic Management: Surgical intervention may be necessary for severe scoliosis, joint contractures, or foot deformities that impair function or cause pain.
- Nutritional Support: Swallowing difficulties may necessitate feeding modifications or gastrostomy tube placement to ensure adequate nutrition and prevent aspiration.
Emerging Therapeutic Approaches
Recent advances in understanding the molecular mechanisms underlying nemaline myopathy have led to the development of potential therapeutic strategies:
- Troponin Activators: Compounds that increase calcium sensitivity of the contractile apparatus, such as tirasemtiv and CK-2127107, have shown promise in preliminary studies by improving muscle function in animal models.
- Gene Therapy: Experimental approaches using viral vectors to deliver functional copies of defective genes are being explored, though significant challenges remain in targeting skeletal muscle effectively.
- Pharmacological Interventions: Small molecules targeting specific pathways involved in muscle development and maintenance are under investigation, including compounds that modulate autophagy and protein quality control mechanisms.
Prognosis and Quality of Life
The prognosis for individuals with nemaline myopathy varies considerably based on the specific genetic subtype and severity of presentation. Patients with severe congenital forms face significant challenges, including respiratory failure and feeding difficulties, which can impact long-term survival. However, with appropriate supportive care, many patients can achieve reasonable quality of life and normal or near-normal lifespan.
Those with milder forms typically have better prognoses, with many achieving independent ambulation and productive lives. The slowly progressive nature of most forms allows for adaptation and the implementation of supportive interventions as needed.
Future Directions
Research into nemaline myopathy continues to advance our understanding of muscle biology and potential therapeutic targets. Areas of active investigation include:
- Biomarker Development: Identification of reliable biomarkers for disease progression could facilitate clinical trials and improve patient monitoring.
- Precision Medicine: Development of mutation-specific therapies based on the underlying genetic defect may offer more targeted and effective treatments.
- Regenerative Medicine: Stem cell-based approaches and tissue engineering strategies may eventually provide options for replacing damaged muscle tissue.
- Clinical Trial Networks: International collaboration through organizations like the Congenital Muscle Disease International Registry is essential for conducting meaningful clinical trials in this rare disease population.
Conclusion
Nemaline myopathy represents a paradigm for understanding rare genetic muscle disorders, demonstrating how advances in molecular genetics, histopathology, and clinical management can improve outcomes for patients with complex neuromuscular conditions. While significant challenges remain in developing effective treatments, the increasing understanding of disease mechanisms and the development of potential therapeutic approaches offer hope for improved management and ultimately, targeted treatments for this challenging condition.
Continued research efforts, international collaboration, and patient advocacy will be essential in advancing our knowledge and developing effective interventions for individuals and families affected by nemaline myopathy. The complexity of this disorder underscores the importance of multidisciplinary care and the need for specialized expertise in managing patients with rare neuromuscular conditions.
This article represents current understanding of nemaline myopathy based on available research and clinical experience. As our knowledge continues to evolve, recommendations for diagnosis and management may change. Patients and families should consult with qualified medical professionals for personalized advice regarding specific clinical situations.
Observer Voice is the one stop site for National, International news, Sports, Editor’s Choice, Art/culture contents, Quotes and much more. We also cover historical contents. Historical contents includes World History, Indian History, and what happened today. The website also covers Entertainment across the India and World.